

Key Points
KIR3DL2, with expression triggered by HTLV-1 infection, is a novel diagnostic marker of acute-type ATL.
The lacutamab monoclonal anti-KIR3DL2 antibody may be an effective strategy to treat ATL and is under study for relapsed/refractory PTCL.

Abstract
Adult T-cell leukemia (ATL) is a lymphoid neoplasm caused by human T-cell leukemia virus type 1 (HTLV-1), which encodes the transcriptional activator Tax, which participates in the immortalization of infected T cells. ATL is classified into 4 subtypes: smoldering, chronic, acute, and lymphoma. We determined whether natural killer receptors (NKRs) were expressed in ATL. NKR expression (KIR2DL1/2DS1, KIR2DL2/2DL3/2DS2, KIR3DL2, NKG2A, NKG2C, and NKp46) was assessed in a discovery cohort of 21 ATL, and KIR3DL2 was then assessed in 71 patients with ATL. KIR3DL2 was the only NKR among those studied frequently expressed by acute-type vs lymphoma- and chronic/smoldering-type ATL (36 of 40, 4 of 16, and 1 of 15, respectively; P = .001), although acute- and lymphoma-type ATL had similar mutation profiles by targeted exome sequencing. The correlation of KIR3DL2 expression with promoter demethylation was determined by microarray-based DNA methylation profiling. To explore the role of HTLV-1, KIR3DL2 and TAX messenger RNA (mRNA) expression levels were assessed by PrimeFlow RNA in primary ATL and in CD4+ T cells infected with HTLV-1 in vitro. TAX mRNA and KIR3DL2 protein expressions were correlated on ATL cells. HTLV-1 infection triggered KIR3DL2 by CD4+ cells but Tax alone did not induce KIR3DL2 expression. Ex vivo, autologous, antibody-dependent cell cytotoxicity using lacutamab, a first-in-class anti-KIR3DL2 humanized antibody, selectively killed KIR3DL2+ primary ATL cells ex vivo. To conclude, KIR3DL2 expression is associated with acute-type ATL. Transcription of KIR3DL2 may be triggered by HTLV-1 infection and correlates with hypomethylation of the promoter. The benefit of targeting KIR3DL2 with lacutamab is being further explored in a randomized phase 2 study in peripheral T-cell lymphoma, including ATL (registered on https://clinicaltrials.gov as #NCT04984837).
Introduction
Adult T-cell leukemia (ATL, a fatal malignant disorder of CD4+ T lymphocytes) is caused by infection with human T-cell leukemia virus type 1 (HTLV-1). It is not clear why a small proportion of HTLV-1–infected individuals develop a clinically aggressive type of ATL. ATL can be classified into 4 clinical subtypes.1 Aggressive forms are divided into acute or lymphoma types, depending on the presence of abnormal lymphocytosis, whereas patients with the chronic or smoldering types are categorized as having indolent ATL. Indolent ATL generally progresses slowly, and patients are carefully monitored by watchful waiting. Chemotherapy is applied if transformation to an aggressive type occurs. However, patients with the chronic or smoldering type could present various symptoms and are offered intensive treatments.2 Little is known about the viral or host molecular determinants of ATL.
HTLV-1 encodes the transcriptional activator Tax, which activates various transcriptional pathways, such as cyclic adenosine monophosphate response element binding protein (CREB) and NF-κB, and it represses p53 and interferes with several cell cycle regulators, including cyclins and cdk inhibitors.3,4 These multiple functions are believed to participate in the immortalization of HTLV-1–infected cells. In contrast, the role of Tax in advanced ATL is unclear because the activator cannot be detected in cells from most patients with ATL,5 probably because of negative selection by the strong anti-Tax cytotoxic T-lymphocyte (CTL) response,6 but may be expressed in vitro in the absence of cytotoxic cells. Furthermore, provirus sense expression in cells is inactivated (by methylation of the 5′ long terminal repeat (LTR), mutation of TAX, or by deletion of the 5′ LTR proviral DNA) in more than half of all patients with ATL.7-10 Recently, 2 reports described high-throughput RNA sequencing techniques to analyze HTLV-1 viral expression; both confirmed that sense RNA expression was absent in >90% of ATL samples.11,12
KIR3DL2 (also known as CD158K) is a killer immunoglobulinlike receptor (KIR) normally expressed by a subset of NK cells and a small proportion of CD4+ and CD8+ T lymphocytes. The receptor’s expression is aberrant in Sézary syndrome and in other cutaneous T-cell lymphomas (CTCLs).13 It has been suggested that in a small proportion of patients with ATL, circulating tumor cells also express KIR3DL2.14 The initial report was recently confirmed by an immunohistochemical assessment of 12 additional patients with ATL.15 In patients with SS, the detection of KIR3DL2 expression on circulating tumor cells is useful for diagnosis, prognosis, and follow-up of tumor cell burden.16-20 Moreover, lacutamab, a humanized cytotoxic monoclonal antibody (mAb) against KIR3DL2, has shown antitumor activity in preclinical studies.21 Lacutamab was well tolerated and had beneficial clinical activity for relapsed/refractory CTCL in a phase 1 study (registered on https://clinicaltrials.gov as #NCT02593045).22
Given the unmet therapeutic needs of patients with ATL, we studied the expression of NK receptors (NKRs), particularly KIR3DL2, in a cohort of patients with ATL and tested the efficacy of lacutamab on primary ATL cells. We also sought to determine the mechanisms of KIR3DL2 expression.
Methods
Patients and quantification of the HTLV-1 proviral load
The study was approved by an independent ethics committee (CPP Ile de France II, CNIL: number 1692254, registration number 000001072), and all living patients gave their written informed consent. The study population’s demographic and clinical characteristics are summarized in supplemental Table 1 (available on the Blood Web site). Clinical data were retrospectively collected. Samples were collected at the time of diagnosis and, depending on the patient, at various times in the course of the disease. Except for 1 patient with Kikuchi disease, all HTLV-1 carriers were asymptomatic. The HTLV-1 plasma viral load (PVL) was quantified with real-time polymerase chain reaction (qRT-PCR) and primers specific for the pX region, as previously described.12,23 The PVL was expressed in HTLV-1 proviral copies per 100 peripheral blood mononuclear cells (PBMCs).
Immunohistochemistry and flow cytometry
Tissues sections were stained with hematoxylin and eosin reagent. Expression of KIR3DL2 was assessed by immunohistochemistry (IHC), using the specific anti-KIR3DL2 mAb clones MOG1-MK323-12B11 and H5 at a dilution of 10 μg/mL (Innate Pharma, Marseille, France), allowing for staining of frozen and formalin-fixed, paraffin-embedded tissue sections, respectively. All immunostainings were performed with an automated Leica Biosystem Bond III system. Flow cytometry (FC) of lymphocytes was performed with 8-color mixes (supplemental Methods). KIR3DL2 and NKp46 were respectively detected with the anti-KIR3DL2 mAb clone 13E4 and the anti-NKp46 mAb clone 9E2 (both provided by Innate Pharma) conjugated to phycoerythrin. The ratio of fluorescence intensity was assessed to determine the positivity of KIR3DL2. Data were analyzed on a FACSCanto II cytometer with FlowJo software (version 10.2, Ashland, OR; Becton Dickinson and Company).
qRT-PCR, targeted exome sequencing, and methylation
KIR3DL2 messenger RNA (mRNA) expression was detected using the Roche Real-Time Ready Assay (ID 136820; supplemental Methods). TAX mRNA was quantified using the TaqMan method according to a previously described protocol.24 Targeted exome sequencing was performed as described previously.9 A microarray-based DNA methylation analysis of samples from 28 patients with ATL, 4 HTLV-1 asymptomatic carriers, and isolated CD4+ T lymphocytes from 4 healthy donors was performed with the Infinium HumanMethylation450 BeadChip system (Illumina, Inc., San Diego, CA; supplemental Methods). Cell lines (PBMCs from healthy donors and patients with ATL), were treated with 5-azacytidine (5-aza; Sigma-Aldrich) and analyzed for KIR3DL2 expression (using FC) after a 72-hour incubation. Direct KIR3DL2 promoter methylation levels were analyzed by Methylation Sensitive-Multiple Ligation-Dependent Probe Assay with a custom probe (supplemental Methods), SALSA MLPA P200 Reference-1 probemix, and EK1 reagent kits from MRC-Holland, according to the manufacturer’s recommendations. Data were analyzed with Coffalyser software (MRC-Holland).
Cell lines, primary ATL cells, and cell culture
The TIB-161 HuT 78 KIR3DL2+ and the TIB-152 Jurkat KIR3DL2− T-cell lines were purchased from the American Type Culture Collection. We also studied the C8166 HTLV-1+ human T cells; the MT-2, MT-4, C91/PL, and HUT-102 HTLV-1 virus-producing T cells; and the TL-OmI ATL–derived cell lines (supplemental Methods). PBMCs from healthy donors and patients with ATL were separated by Ficoll-Hypaque density gradient centrifugation.
HTLV-1 infection in vitro and transfection
PBMCs were cultured alone, stimulated with 1 μg/mL of phytohemagglutinin (Sigma, St Quentin Fallavier, France) and 20 U/mL interleukin-2 (Roche, Boulogne-Billancourt, France), or stimulated and then infected with purified HTLV-1 from an MT-2 culture supernatant. The MT-2 cells were seeded (106 per milliliter) in culture medium and grown overnight. The supernatant was collected and centrifuged for 2 hours at 100 000g. HTLV-1 viruses were quantified using an HTLV-1-p19 ELISA kit (Gentaur). Several concentrations of HTLV-1 (0-900 ng/mL p19 equivalent) were used to stimulate the PBMCs in vitro. T-cell lines were transfected with the DMRIE-C reagent (Roche). The pSG5M empty vector and the pSG5M-Tax plasmids have been described.25 The pGEX-2T and pGEX-2T-Tax (wild-type and m47) plasmids were kindly provided by V. Mocquet (École Normale Supérieure [ENS], Lyon, France), and the pGEX-2T-p24 (GST-CA) plasmid has been described.26
PrimeFlow RNA assay and imaging flow cytometry
The Affymetrix PrimeFlow RNA assay was performed according to the manufacturer’s instructions (supplemental Methods). In brief, the PrimeFlow RNA assay is based on the binding of a specific oligonucleotide probe set to the TAX RNA sequence, followed by hybridization of multiple amplifier molecules for signal amplification and conjugation to a fluorescent dye that can then be quantified by FC. Lymphocytes were identified on the basis of their forward and side scatter properties, and live cells were selected after the exclusion of doublets. CD3− and CD4+ lymphocytes were then assessed for TAX mRNA expression and KIR3DL2 protein expression. Fluorescent signals from antibodies and probes were quantified by FC on an LSR Fortessa system running FACS DIVA software (version 7; BD Biosciences). Images were acquired with the ImageStream system and analyzed on an Imagestream ISX mkII cytometer (Amnis Luminex), which use a combination of FC and cell imaging to analyze a very large number of events. Between 30 000 and 50 000 events were collected in each experiment. Single-stain controls were run for each fluorochrome used, and spectral compensation was performed with IDEAS software (version 6.2; Amnis Luminex). Specific masks were designed for the analysis of nuclear or cytoplasmic localizations of KIR3DL2 and TAX mRNA in live cells.
Ex vivo autologous antibody-dependent cellular cytotoxicity assays
Autologous antibody-dependent cellular cytotoxicity (ADCC) assays were performed on isolated cells, using the indicated mAbs. PBMCs were split into 2 samples, and CD4+ T cells and autologous NK cells were purified (using negative isolation by magnetic bead labeling) with the respective MACS isolation kits (Miltenyi Biotech). Isolated CD4+ T cells were preincubated for 30 minutes at room temperature with the anti-KIR3DL2 lacutamab (provided by Innate Pharma), the negative control (IC), or the positive control (the anti-CD52 alemtuzumab), all at 20 μg/mL, and then mixed with autologous NK lymphocytes at the indicated effector/target (E/T) ratios. The initial E/T ratio was determined by FC. The cells were incubated for 4 to 6 hours at 37°C in RPMI/10% fetal bovine serum. The death of ATL cells was monitored by FC after incubation with 7-aminoactinomycin D (7-AAD) cell viability stain (eBioscience).
Statistical analysis
Continuous variables were quoted as the median (interquartile range), and groups were compared by unpaired t test. Confidence intervals were estimated with a continuity correction. The patients’ tumor cells were defined as KIR3DL2+ or KIR3DL2− as assessed by IHC and/or FC, to define 2 groups. Survival in each of the groups of patients was estimated according to the Kaplan-Meier method, and the groups were compared by using the log-rank test. All graphic and statistical analyses were performed with GraphPad Prism software (version 7, San Diego, CA) and R software.27 The threshold for statistical significance was set to P < .05.
Results
KIR3DL2 expression is associated with acute-type ATL
Expression of NKRs (KIR2DL1/2DS1, KIR2DL2/2DL3/2DS2, KIR3DL2, NKG2A, NKG2C, and NKp46) was measured on PBMCs from a discovery cohort of 21 patients with ATL, using FC. ATL cells were identified by their CD4 and low CD3/TCRαβ expression and the absence of CD7 expression (supplemental Figure 1). KIR3DL2 was the only NKR expressed on CD4+ CD7-ATL tumor cells among those studied (Figure 1A). In total, KIR3DL2 expression was next assessed in 110 samples collected from 71 patients with ATL and 8 HTLV-1 asymptomatic carriers (ACs) by FC and/or IHC (supplemental Table 1; supplemental Figure 2). Of the 40 patients with acute-type ATL, 36 (90%) had KIR3DL2+ abnormal lymphocytes with a median of 53% of KIR3DL2+ cells (Figure 1C). In contrast, KIR3DL2 positivity was much less frequent among patients with lymphoma or chronic/smoldering types of ATL (4 of 16 and 1 of 15, respectively; Figure 1B-C). Moreover, the KIR3DL2− patient with an initial diagnosis of chronic-type ATL became KIR3DL2+ after transformation to the acute type (Figure 1D). KIR3DL2 was not found on CD4+ lymphocytes from HTLV-1 carriers, except 1 patient with Kikuchi disease where a few cells expressed KIR3DL2. We used qRT-PCR KIR3DL2 to assess mRNA expression on PBMCs from 48 patients with ATL and 5 with HTLV-1 ACs. KIR3DL2 mRNA was found in most acute type cases (23 of 30; 74%), but was less frequently present in chronic type (7 of 13; 54%) cases and was absent in lymphoma-type (Figure 1E) cases. In 2 patients with chronic-type ATL and high KIR3DL2 mRNA expression levels, the ATL subsequently transformed into acute-type ATL. KIR3DL2 mRNA levels correlated with KIR3DL2 protein expression (supplemental Figure 3A). In 40 healthy donors, a median of 3% and 9.3% of CD4+ and CD8+ T lymphocytes expressed KIR3DL2, respectively (supplemental Table 2). CD4+CD7− T lymphocytes from 19 healthy donors did not express KIR3DL2, compared with CD4+CD7− from patients with ATL (supplemental Figure 4; P = .0002). Neither protein nor mRNA expression of KIR3DL2 was observed in the HTLV-1–infected (MT-2, C91PL, and C8166) or ATL-derived cell line (TL-OmI and HUT-102).
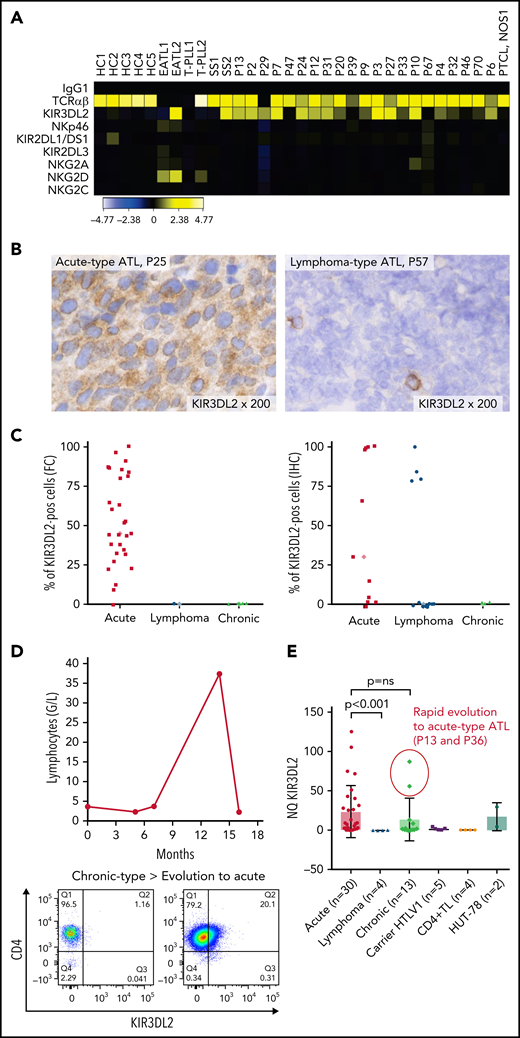
The NK receptor KIR3DL2 is expressed by tumor cells in patients with acute-type ATL. (A) Heat map showing NK receptor expression (defined using FC) in PBMCs sampled from 5 healthy controls (HCs), 2 patients with enteropathy-associated T-cell lymphoma (EATL), 2 patients with T-cell prolymphocytic leukemia (T-LPL), 2 patients with Sezary syndrome (SS), 1 patient with peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS), and 21 patients with ATL. The heat map represents the ratio of fluorescence intensity, as regards the isotypic control (IgG1), expressed as an arbitrary unit, to scale the color code of the heat map using the cytobank software (https://www.cytobank.org). High protein expressions are shown in yellow, and negative protein expressions are shown in black. KIR3DL2 was the only NKR expressed on ATL tumor cells among those studied. (B) IHC assessment of KIR3DL2 protein expression on representative lymph node biopsy specimens (original magnification, ×200), showing that a patient with acute-type ATL (P25) expressed KIR3DL2, whereas a patient with lymphoma-type ATL (P57) did not. (C) Percentage of KIR3DL2+ cells among tumor cells in patients with ATL by FC or IHC, according to the ATL subtype (acute, lymphoma, and chronic type). (D) Example of a patient (P18) with KIR3DL2− chronic-type ATL, which became KIR3DL2+ at the time of the transformation into an acute-type ATL. After 12 months of evolution, the patient presented with lymphocytosis (38.3 × 103 μL) with 96% abnormal KIR3DL2+ lymphocytes, high levels of lactate dehydrogenase, and lymph node and central nervous system involvement, leading to a diagnosis of an acute-type ATL. (E) KIR3DL2 mRNA expression (assessed by qRT-PCR) in PBMCs from 48 patients with ATL, 5 HTLV-1 carriers, CD4+ T lymphocytes from 4 healthy donors, and Hut 78 cells (2-sided Mann-Whitney nonparametric test). Two patients (P13 and P36) with high KIR3DL2 mRNA expression levels were diagnosed as having chronic-type ATL at the time of mRNA analysis; subsequently, the diseased transformed into KIR3DL2+ acute-type ATL.
The NK receptor KIR3DL2 is expressed by tumor cells in patients with acute-type ATL. (A) Heat map showing NK receptor expression (defined using FC) in PBMCs sampled from 5 healthy controls (HCs), 2 patients with enteropathy-associated T-cell lymphoma (EATL), 2 patients with T-cell prolymphocytic leukemia (T-LPL), 2 patients with Sezary syndrome (SS), 1 patient with peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS), and 21 patients with ATL. The heat map represents the ratio of fluorescence intensity, as regards the isotypic control (IgG1), expressed as an arbitrary unit, to scale the color code of the heat map using the cytobank software (https://www.cytobank.org). High protein expressions are shown in yellow, and negative protein expressions are shown in black. KIR3DL2 was the only NKR expressed on ATL tumor cells among those studied. (B) IHC assessment of KIR3DL2 protein expression on representative lymph node biopsy specimens (original magnification, ×200), showing that a patient with acute-type ATL (P25) expressed KIR3DL2, whereas a patient with lymphoma-type ATL (P57) did not. (C) Percentage of KIR3DL2+ cells among tumor cells in patients with ATL by FC or IHC, according to the ATL subtype (acute, lymphoma, and chronic type). (D) Example of a patient (P18) with KIR3DL2− chronic-type ATL, which became KIR3DL2+ at the time of the transformation into an acute-type ATL. After 12 months of evolution, the patient presented with lymphocytosis (38.3 × 103 μL) with 96% abnormal KIR3DL2+ lymphocytes, high levels of lactate dehydrogenase, and lymph node and central nervous system involvement, leading to a diagnosis of an acute-type ATL. (E) KIR3DL2 mRNA expression (assessed by qRT-PCR) in PBMCs from 48 patients with ATL, 5 HTLV-1 carriers, CD4+ T lymphocytes from 4 healthy donors, and Hut 78 cells (2-sided Mann-Whitney nonparametric test). Two patients (P13 and P36) with high KIR3DL2 mRNA expression levels were diagnosed as having chronic-type ATL at the time of mRNA analysis; subsequently, the diseased transformed into KIR3DL2+ acute-type ATL.
The diagnostic value of KIR3DL2 expression was confirmed by its prognostic impact on the whole cohort of patients with ATL. The 2-year overall survival rate was significantly lower in KIR3DL2+ cases (18% vs 52% in KIR3DL2− cases; P < .001; Figure 2A) but was similar in aggressive ATL subtypes according to KIR3DL2 expression (Figure 2B; NS).
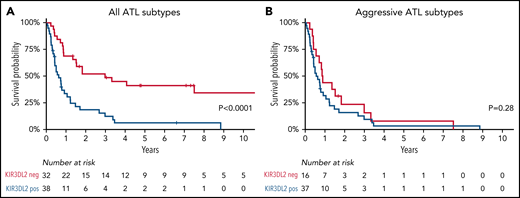
Overall survival. KIR3DL2+ (blue) vs KIR3DL2− (red) patients with all subtypes of ATL (A) and with aggressive ATL subtypes (acute and lymphoma types) (B).
Overall survival. KIR3DL2+ (blue) vs KIR3DL2− (red) patients with all subtypes of ATL (A) and with aggressive ATL subtypes (acute and lymphoma types) (B).
Gene mutations in 40 patients with aggressive subtypes (8 KIR3DL2− and 32 KIR3DL2+ cases) were analyzed with data obtained from previously published targeted exome sequencing.9 There were no significant differences in the mutation profiles between the KIR3DL2− and KIR3DL2+ groups, suggesting that the molecular pathogeneses are similar (supplemental Figure 5). At least 1 activating mutation was found in the TCR-NF-κB pathway, the T-cell trafficking pathway, and the immune escape pathway (n = 6 [75%] vs 27 [85%] and 5 [62%] vs 17 [52%]; and n = 3 [37%] vs 10 [30%] in KIR3DL2− compared with KIR3DL2+cases, respectively), and at least 1 mutation in the cell cycle control pathway was detected in 2 [25%] vs 10 [30%] in KIR3DL2+ cases.
KIR3DL2 expression correlated with KIR3DL2 gene promoter hypomethylation
Given that DNA methylation is known to influence variegated KIR expression in NK cells,28 we assessed the KIR3DL2 promoter’s DNA methylation profile in 28 patients with ATL, 4 HTLV-1 ACs, and 4 healthy donors. The KIR3DL2 promoter was significantly less methylated in acute-type than in lymphoma- and chronic-/smoldering-type ATLs or HTLV-1 ACs; this result was consistent with KIR3DL2 expression (Figure 3A; supplemental Figure 3B). Two patients with lymphoma-type ATL (P57 and P58) had KIR3DL2 promoter hypomethylation but did not express KIR3DL2. Interestingly, they were brothers, suggesting a similar lymphomagenesis in both cases. To determine whether KIR3DL2 transcription was a consequence of DNA hypomethylation, we next measured the induction of KIR3DL2 transcription in the KIR3DL2+ Hut 78 and the KIR3DL2− Jurkat T-cell lines after treatment with the DNA methylase inhibitor 5-aza. After 72 hours of 5-aza treatment, cell-surface KIR3DL2 expression was efficiently and dose-dependently induced on Hut 78 cells but not on Jurkat cells (Figure 3B). Using a custom MS-MLPA assay, we studied the DNA methylation of KIR3DL2 promoter. As expected, KIR3DL2 promoter methylation was significantly lower in acute-type ATL (n = 6; median methylation ratio, 0.66) and Hut 78 cells (methylation ratio, 0.41) compared with normal PBMCs (n = 9; methylation ratio, 0.91) (supplemental Figure 6). Moreover, 5-aza treatment led to a significant decrease (26.2%) in KIR3DL2 methylation from a minimum dose of 1 µM (methylation ratios: 0.31 vs. 0.42) (Figure 3C). By contrast, KIR3DL2 expression on PBMCs from healthy donors and from 4 KIR3DL2+ patients with ATL did not increase upon ex vivo 5-aza treatment. Last, CD4+ T cells from 2 patients treated with 5-aza in vivo for myelodysplastic syndrome did not express KIR3DL2. These results suggest that in ATL cells, KIR3DL2 protein expression may be maintained by DNA hypomethylation at the KIR3DL2 promoter. However, DNA-demethylating treatment alone does not induce KIR3DL2 expression in a negative baseline state and does not increase the demethylation status of the KIR3DL2 promoter in acute-type ATL.
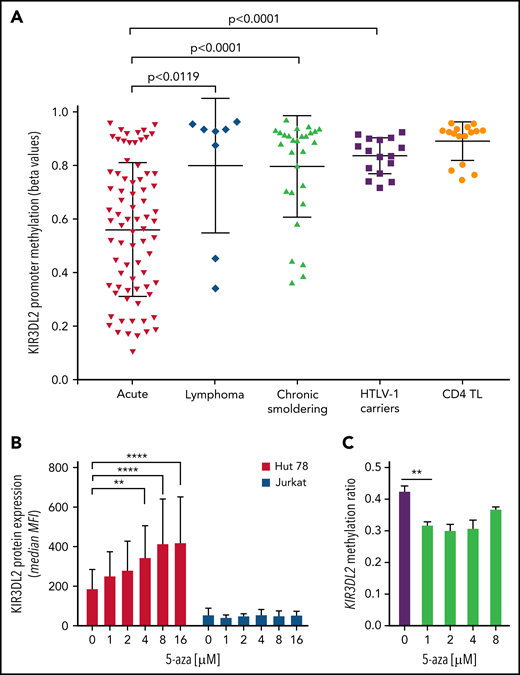
KIR3DL2 promoter is hypomethylated in patients with acute-type ATL. (A) Differential methylation at the KIR3DL2 locus using the β-value method, measured in an Illumina 450K array-based methylome analysis in samples from patients with ATL (19 acute, 2 lymphoma, and 7 chronic/smoldering forms), 4 HTLV-1 carriers, and 4 healthy donors (isolated CD4+ TL). The KIR3DL2 promoter was significantly less methylated in acute-type ATL than in lymphoma- and chronic/smoldering-type ATLs or HTLV-1 ACs (unpaired t test). The proportion of tumor cells was greater than 85% in the acute-type ATL samples. Each dot represents an individual CpG per patient in the β-value analysis. The horizontal line represents the median, and the whiskers represent the range. (B) KIR3DL2 protein expression was induced on Hut 78 cells, but not on Jurkat cells, upon incubation with 5-aza (0, 1, 2, 4, 8, or 16 μM) for 3 days. The error bars correspond to the standard deviation. ****P < .0001; **P < .01 (2-way analysis of variance followed by Šidák’s post hoc test). MFI, mean fluorescence intensity. (C) 5-Aza led to hypomethylation of KIR3DL2 promoter, assessed by MS-MLPA, in Hut 78 cells treated with 5-aza from 1 μM. Experiment performed in triplicate. **P < .01 (unpaired t-test).
KIR3DL2 promoter is hypomethylated in patients with acute-type ATL. (A) Differential methylation at the KIR3DL2 locus using the β-value method, measured in an Illumina 450K array-based methylome analysis in samples from patients with ATL (19 acute, 2 lymphoma, and 7 chronic/smoldering forms), 4 HTLV-1 carriers, and 4 healthy donors (isolated CD4+ TL). The KIR3DL2 promoter was significantly less methylated in acute-type ATL than in lymphoma- and chronic/smoldering-type ATLs or HTLV-1 ACs (unpaired t test). The proportion of tumor cells was greater than 85% in the acute-type ATL samples. Each dot represents an individual CpG per patient in the β-value analysis. The horizontal line represents the median, and the whiskers represent the range. (B) KIR3DL2 protein expression was induced on Hut 78 cells, but not on Jurkat cells, upon incubation with 5-aza (0, 1, 2, 4, 8, or 16 μM) for 3 days. The error bars correspond to the standard deviation. ****P < .0001; **P < .01 (2-way analysis of variance followed by Šidák’s post hoc test). MFI, mean fluorescence intensity. (C) 5-Aza led to hypomethylation of KIR3DL2 promoter, assessed by MS-MLPA, in Hut 78 cells treated with 5-aza from 1 μM. Experiment performed in triplicate. **P < .01 (unpaired t-test).
TAX mRNA expression corelates with KIR3DL2 protein expression in primary ATL cells
We used the PrimeFlow RNA assay and KIR3DL2 immunolabeling to assess the correlation between TAX mRNA expression and KIR3DL2 protein expression in primary ATL cells from 3 patients with acute-type disease (P9, P18, and P28). For 2 patients, analyses were performed using the ImageStream imaging flow cytometer. It is noteworthy that although Tax expression is weak in fresh primary ATL cells, nearly 50% of cells from patients with ATL express viral antigens after ex vivo culture.29 Hence, to induce TAX expression, we cultured primary ATL cells for 12 to 15 hours before the PrimeFlow RNA assay. On ATL cells, TAX mRNA expression correlated with KIR3DL2 protein expression (Figure 4; supplemental Figure 7), with a cytoplasmic localization (Figure 4B). These results suggest that infected cells could preferentially express KIR3DL2.
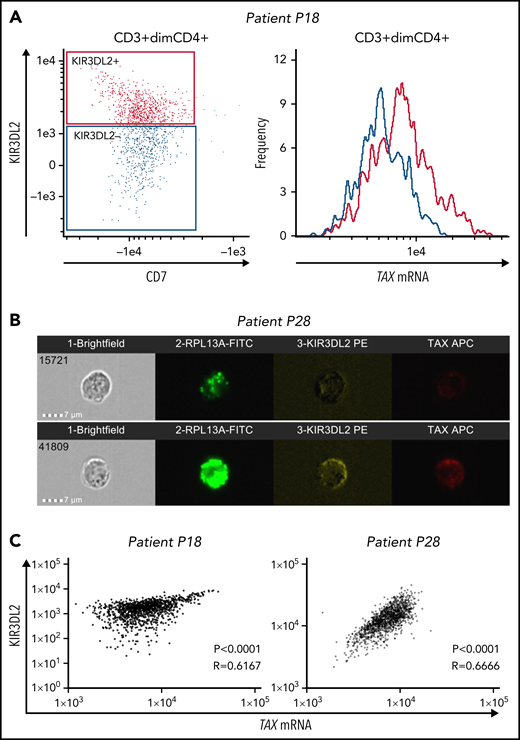
TAX mRNA and KIR3DL2 protein expressions correlate in primary ATL tumor cells. Primary ATL cells from 3 patients were cultured in vitro for 12 to 15 hours before the PrimeFlow RNA assay. For 2 of the patients (P18 and P28), TAX mRNA expression and KIR3DL2 cell surface protein expression were quantified with the ImageStream X Mark II imaging flow cytometer. (A) The expression of TAX mRNA was higher in tumor cells expressing KIR3DL2 (red) than in those not expressing it (blue). (B) Two illustrative images of ImageStream experiments. Cells were probed for the positive control RPL13A, KIR3DL2, and TAX mRNA expression and run on the ImageStream. Bar represents 7 μm. Tumor cells coexpress KIR3DL2 and TAX mRNA with a cytoplasmic localization or express neither KIR3DL2 nor TAX mRNA. (C) Correlation between TAX mRNA expression and KIR3DL2 cell surface protein expression quantified with the ImageStream for 2 of the patients (P18 and P28). The P value and Spearman correlation coefficient are indicated.
TAX mRNA and KIR3DL2 protein expressions correlate in primary ATL tumor cells. Primary ATL cells from 3 patients were cultured in vitro for 12 to 15 hours before the PrimeFlow RNA assay. For 2 of the patients (P18 and P28), TAX mRNA expression and KIR3DL2 cell surface protein expression were quantified with the ImageStream X Mark II imaging flow cytometer. (A) The expression of TAX mRNA was higher in tumor cells expressing KIR3DL2 (red) than in those not expressing it (blue). (B) Two illustrative images of ImageStream experiments. Cells were probed for the positive control RPL13A, KIR3DL2, and TAX mRNA expression and run on the ImageStream. Bar represents 7 μm. Tumor cells coexpress KIR3DL2 and TAX mRNA with a cytoplasmic localization or express neither KIR3DL2 nor TAX mRNA. (C) Correlation between TAX mRNA expression and KIR3DL2 cell surface protein expression quantified with the ImageStream for 2 of the patients (P18 and P28). The P value and Spearman correlation coefficient are indicated.
HTLV-1 infection, but not Tax expression alone, induces KIR3DL2 expression on CD4+ T cells
To explore the role of HTLV-1 infection in KIR3DL2 expression, we assessed PBMCs obtained from healthy donors that had been activated or infected with HTLV-1 in vitro. Purified HTLV-1 virions induced KIR3DL2 expression by CD4+ T cells in a dose-dependent manner (ie, p19 equivalent, n = 3; Figure 5A; supplemental Figure 8). The induced KIR3DL2 expression was observed after 7 days of cell culture but not after 24 hours (supplemental Figure 9). The HTLV-1 PVL was quantifiable in infected PBMCs, which attests to the existence of reverse transcription and (probably) HTLV-1 integration. Likewise, KIR3DL2 expression was not induced by activation of CD4+ T cells with phytohemagglutinin/interleukin-2, suggesting that a stable infection (rather than activation alone) is necessary for KIR3DL2 expression (supplemental Figure 10). It is noteworthy that this effect was restricted to KIR3DL2, because neither KIR3DS1 nor NKp46 was induced by HTLV-1 infection (supplemental Figure 11). To confirm the specificity of our data, we applied the PrimeFlow RNA assay and immunolabeling of KIR3DL2. As expected, TAX mRNA was induced by HTLV-1 infection in a dose-dependent manner. KIR3DL2 protein and TAX mRNA expression correlated on CD4+ lymphocytes. Moreover, TAX mRNA was mostly expressed in KIR3DL2+ CD4+ lymphocytes, whereas KIR3DL2− CD4+ lymphocytes were also negative for TAX mRNA (Figure 5B). We then hypothesized that Tax itself may induce KIR3DL2 expression. We therefore decided to transfect Jurkat T cells and use qRT-PCT to measure mRNA levels of KIR3DL2 and the positive control ICAM-1. After 24 hours, overexpression of Tax led to an increase in ICAM-1 transcripts but not KIR3DL2 expression (supplemental Figure 12). Moreover, among the informative patients with ATL, 35% (6 of 17) expressed KIR3DL2, despite having a deletion of the 5′-LTR region that carries the viral promoter, and therefore could not express Tax. Finally, 2 patients (P3 and P36) and 1 HTLV-1 carrier (C1) did not express TAX mRNA by qRT-PCR, even though they expressed the KIR3DL2 protein (supplemental Table 3). Taken together, these results indicate that the presence of Tax alone does not induce KIR3DL2 mRNA expression.
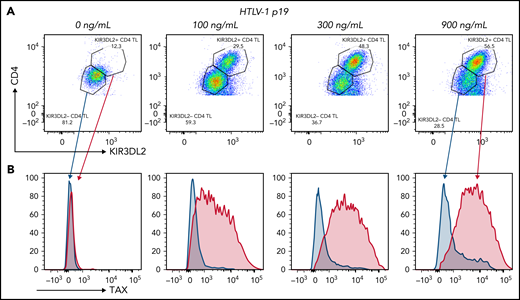
HTLV-1 infection in vitro induced the expression of KIR3DL2 by CD4+ T cells (representative assay). (A) Cell-free HTLV-1 virions induced the expression of KIR3DL2 by CD4+ T cells after 7 days of culture. PBMCs from healthy donors were cultured with increasing concentrations of HTLV-1 (p19-equivalent), and KIR3DL2 cell surface expression was quantified by FC. Purified HTLV-1 virions induced KIR3DL2 expression by CD4+ T cells in a dose-dependent manner. (B) TAX mRNA was mostly expressed by KIR3DL2+ CD4+ lymphocytes and was not observed in KIR3DL2− CD4+ lymphocytes, quantified using the PrimeFlow RNA assay and KIR3DL2 immunolabeling.
HTLV-1 infection in vitro induced the expression of KIR3DL2 by CD4+ T cells (representative assay). (A) Cell-free HTLV-1 virions induced the expression of KIR3DL2 by CD4+ T cells after 7 days of culture. PBMCs from healthy donors were cultured with increasing concentrations of HTLV-1 (p19-equivalent), and KIR3DL2 cell surface expression was quantified by FC. Purified HTLV-1 virions induced KIR3DL2 expression by CD4+ T cells in a dose-dependent manner. (B) TAX mRNA was mostly expressed by KIR3DL2+ CD4+ lymphocytes and was not observed in KIR3DL2− CD4+ lymphocytes, quantified using the PrimeFlow RNA assay and KIR3DL2 immunolabeling.
In an ex vivo assay, lacutamab efficiently enables autologous NK cells to eliminate KIR3DL2+ primary ATL cells
Lacutamab is a first-in-class anti-KIR3DL2 humanized cytotoxicity-inducing antibody that was selected for its potent antitumor effector functions. It is currently in clinical trials for treatment of CTCL. We evaluated lacutamab’s antitumor efficacy by using tumor cells from patients with ATL as targets (T) and autologous NK cells as effectors (E) (gating strategy is shown in supplemental Figure 13; supplemental Table 4). This autologous model is relevant because as it takes into account both the functional status of NK cells in patients with ATL and the intrinsic sensitivity of tumor cells to lacutamab-mediated ADCC. Activity of lacutamab against primary ATL cells was observed in all 4 of the KIR3DL2+ patient samples tested, and killing of cells increased with the E/T ratio. In most cases, lacutamab’s anti–tumor cell effect was evidenced at an E/T ratio of 1:1 or more, with significant killing of ATL cells, compared with the negative control condition at an E/T ratio of 5:1 (P = .0006; Figure 6A; supplemental Figure 14). Regardless of the E/T ratio, nonspecific (baseline) NK-mediated cytotoxicity was negligible, as shown by the absence of greater killing with the IC antibody. Moreover, lacutamab did not mediate the killing of KIR3DL2− ATL cells in patients with KIR3DL2+ or KIR3D3L2− ATL (Figure 6B-D). We used the same assay to show that lacutamab antibodies did not induce the death of effector NK cells from patients with ATL (Figure 6C; supplemental Figure 15). Finally, we confirmed these results with ADCC experiments on frozen KIR3DL2+ primary ATL cells (heterologous NK cells from healthy donors; n = 3; supplemental Figure 16). These observations suggest that (1) primary KIR3DL2+ ATL cells are selectively sensitive to ADCC mediated by lacutamab through KIR3DL2 targeting, and (2) NK cells from patients with ATL are functional and can mediate potent lacutamab-driven ADCC.
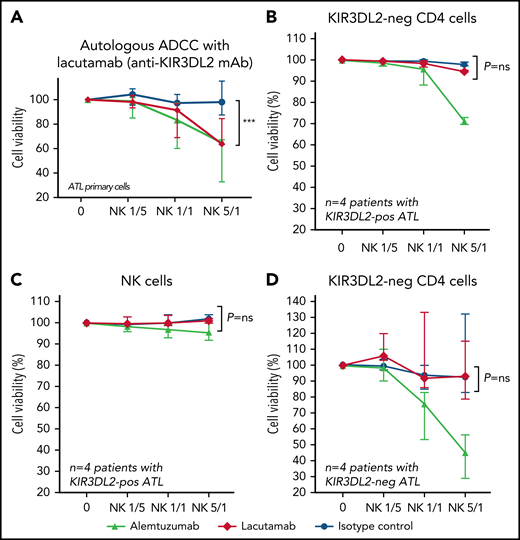
Lacutamab specifically eliminates KIR3DL2-positive primary ATL cells in an autologous ADCC assay. CD4+ T cells from patients with KIR3DL2-positive ATL (n = 4) or patients with KIR3DL2-negative ATL (n = 4) were preincubated with alemtuzumab (triangles, positive control), isotype control (circles, negative control), or the anti-KIR3DL2 mAb lacutamab (red diamonds). After the addition of autologous NK cells at the indicated E/T ratio, the death of KIR3DL2+ (A) and KIR3DL2− (B) CD4+ tumor cells, and NK cells (C) of patients with KIR3DL2+ ATL was monitored using 7-AAD cell viability stain. As control, the death of KIR3DL2− tumor cells in patients with KIR3DL2− ATL were also assessed (D). The results are expressed as the proportion of cell viability in each cellular population at a given E/T ratio. The cellular populations that were assessed are indicated above each plot. ***P < .001, ns, not significant (two-way ANOVA followed by Sidak’s post-test).
Lacutamab specifically eliminates KIR3DL2-positive primary ATL cells in an autologous ADCC assay. CD4+ T cells from patients with KIR3DL2-positive ATL (n = 4) or patients with KIR3DL2-negative ATL (n = 4) were preincubated with alemtuzumab (triangles, positive control), isotype control (circles, negative control), or the anti-KIR3DL2 mAb lacutamab (red diamonds). After the addition of autologous NK cells at the indicated E/T ratio, the death of KIR3DL2+ (A) and KIR3DL2− (B) CD4+ tumor cells, and NK cells (C) of patients with KIR3DL2+ ATL was monitored using 7-AAD cell viability stain. As control, the death of KIR3DL2− tumor cells in patients with KIR3DL2− ATL were also assessed (D). The results are expressed as the proportion of cell viability in each cellular population at a given E/T ratio. The cellular populations that were assessed are indicated above each plot. ***P < .001, ns, not significant (two-way ANOVA followed by Sidak’s post-test).
Discussion
In a retrospective study, we showed that KIR3DL2 expression is a useful biomarker for identifying acute-type ATL. Furthermore, our data suggest that KIR3DL2 expression by ATL cells may be induced by HTLV-1 infection and regulated by DNA hypomethylation of the gene’s promoter. Last, our results suggest that KIR3DL2 may be a novel therapeutic target in select patients with ATL.
It was recently demonstrated that high KIR3DL2 expression is a characteristic feature of Sézary syndrome.17-19 Furthermore, marked KIR3DL2 expression has been observed in all CTCL13 and in other PTCL subtypes (see Decroos et al30). We showed that KIR3DL2 expression is also a hallmark of acute-type ATL in all involved organs. Moreover, KIR3DL2 expression was associated with a low survival rate, which is in line with the poor prognosis observed for acute-type ATL.31 Because KIR3DL2 can be detected easily and specifically on routinely fixed tissue sections and using FC, this is a robust marker that facilitates the diagnosis of acute-type ATL. ATL cells usually express CD3, CD4, and CD25, but not CD7.32 The ectopic expression of TSLC1/CADM1 may constitute a biomarker for HTLV-1–infected cells and acute-type ATL.33,34 Although the loss of CD7 in the CADM1+ subpopulation is associated with the onset of aggressive ATL,35,36 a positive marker of acute-type ATL has not been identified. Despite the occasional presence of KIR3DL2+ cells in lymphoma- and chronic-type ATL, KIR3DL2 expression may be a marker of acute-type ATL or transformation of chronic-type ATL.
KIR expression is clonally distributed in NK cells, but can also be acquired in T lymphocytes after chronic antigenic stimulation, after exposure to cytokines,37,38 or after malignant transformation. However, the underlying mechanisms have yet to be characterized.39 In our study, tumor cells from patients with acute- or lymphoma-type ATL had the same mutational profile, suggesting that the molecular pathogenesis was similar. Furthermore, DNA hypomethylation of the KIR3DL2 promoter was found to correlate with KIR3DL2 protein expression in ATL cells. In vitro treatment with the methyltransferase inhibitor 5-aza led to a decrease of KIR3DL2 promoter methylation and was associated with greater KIR3DL2 expression in a KIR3DL2+ cell line but not in KIR3DL2− cells. Although activation of CD4+ T cells by phytohemagglutinin/interleukin-2 did not induce the surface expression of KIR3DL2 protein, HTLV-1 infection triggered KIR3DL2 expression by normal CD4+ cells under the conditions of the experiments and after 7 days of cell culture, suggesting that a stable infection (rather than activation alone) is required for KIR3DL2 expression. It remains to be determined whether HLTV-1 induces KIR3DL2 transcription in all CD4+ cells or solely in a preexisting subpopulation of KIR3DL2+ CD4+ T cells. Indeed, the origin of the ATL cells is subject to debate,40 and a small population of KIR3DL2+ CD4 T cells was identified in healthy donors. However, this effect is also observed in vitro in KIR3DL2− CD4+ T cells, and so the “subpopulation hypothesis” is unlikely; it is more likely that HTLV-1 influences KIR3DL2 expression either through infection-dependent chronic antigenic stimulation, viral integration, or the transcriptional activity of viral proteins. We evidenced a correlation between TAX mRNA expression and KIR3DL2 protein expression in HTLV-1–infected CD4+ cells in vitro and in primary ATL cells ex vivo. Tax may therefore have a role in the induction of KIR3DL2 expression. The KIR3DL2 gene promoter contains binding sites for NF-κB and cyclic adenosine monophosphate response element binding protein, 2 transcription factors known to be activated by Tax.3 However, Tax alone is not sufficient to induce KIR3DL2 expression. Taken as a whole, these results suggest that KIR3DL2 gene transcription may be induced by HTLV-1 infection and regulated by DNA hypomethylation of the gene promoter. Such a 2-step process may explain why KIR3DL2 expression was restricted to acute-type ATL, even though HTLV-1 was integrated into all ATL subtypes.
The international consensus on the treatment of ATL is based on the clinical subtype and prognostic factors.41,42 Treatment options include intensive chemotherapy; allogeneic hematopoietic stem cell transplantation31; a combination of arsenic trioxide, interferon α, and zidovudine (for chronic-type ATL)43,44; and mogamulizumab for relapsed/refractory CCR4+ ATL.45 Nevertheless, the prognosis for patients with ATL remains poor.31,46 Acute-type cases of ATL are particularly refractory to chemotherapy, and patients often relapse before transplantation.47,48 In proof-of-concept experiments, we showed in this study that lacutamab, an anti-KIR3DL2 humanized mAb currently in clinical trials for CTCL, was effective in autologous killing assays ex vivo, where it induces NK-cell–based lysis of primary ATL cells. The efficacy of lacutamab against primary ATL cells was similar to what has been observed in previous similar ADCC assays against primary Sezary cells.21 These results demonstrated both the functional status of the ATL patients’ NK cells and the sensitivity of primary ATL cells to ADCC via KIR3DL2 targeting. As previously described, we did not find that lacutamab reduces the count of KIR3DL2+ NK cells.22 Our present data therefore provide the preclinical rationale for considering the clinical use of lacutamab in patients with ATL. In this respect, the benefit of targeting KIR3DL2 with lacutamab is being further explored in a randomized phase 2 study in relapsed/refractory peripheral T-cell lymphoma, including ATL (registered on https://clinicaltrials.gov at #NCT04984837).
In summary, our present results demonstrated that KIR3DL2 is a marker with diagnostic value for acute-type ATL. The induction of KIR3DL2 by HTLV-1 infection revealed a previously unknown trigger of KIR protein expression in CD4+ cells. Further studies of the role of KIR3DL2 in lymphomagenesis are now warranted. Lacutamab is currently in development in advanced CTCL (registered on http://clinicaltrials.gov as #NCT03902184),22 and our findings open up the way to a novel treatment for acute-type ATL that will be included in the randomized phase 2 study “KILT,” evaluating lacutamab in patients with relapsed/refractory KIR3DL2+ PTCL (#NCT04984837).
Acknowledgments
The authors thank Innate Pharma for providing the anti-KIR3DL2 antibodies used for IHC, FC, and ADCC experiments.
This study was supported by INSERM (O.H., M.C.), INSERM Plan Cancer 2009-2013 (C.P.), Innate Pharma (Marseille, France) (M.C., J.B., O.H.), Institut National du Cancer (INCA) (M.C.), la Ligue Contre le Cancer (O.H.), l’Association pour la Recherche Contre le Cancer (O.H.), and Université de Paris Cité (M.C., J.B., O.H.). M.C. is the recipient of a grant from INCA.
Authorship
Contribution: M.C. and O.H. conceptualized and supervised the study and wrote the manuscript; M.C., J.B., L.L., A.M., V.A., P.P., V.A.-F., M.D., Y.L., C.P., and L.W. provided data and data analysis; M.C. contributed to statistical interpretation; M.C., J.B., L.L., L.F., F.S., L.N., C.B., C.P., V.A., A.M., and O.H. provided patient material and clinical data; M.C., J.B., and O.H. obtained the funding; H.S., A.T., E.B. and L.G. to provided data and data analysis; N.O. and P.G. to provided patient material and clinical data; all authors edited and approved the final version of the manuscript; and M.C. and O.H. were responsible for the final version of the manuscript.
Conflict-of-interest disclosure: F.L. and C.B, are senior directors of and have ownership interest (including patents) in Innate Pharma. M.C., J.B., N.O., P.G., and O.H. received research grants from Innate Pharma. The remaining authors declare no competing financial interests.
Correspondence: Olivier Hermine, Department of Clinical Hematology, Necker Hospital, 149 rue de Sèvres, 75015 Paris, France; e-mail: ohermine@gmail.com.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal