

Key Points
Helper T-cell response to coagulation factor VIII critically depends on a select set of anatomically distinct antigen-presenting cells.
Innate immune signaling and an antigen trafficking pattern that skirts red pulp macrophages shape the immunogenicity of FVIII.

Abstract
Despite >80 years of clinical experience with coagulation factor VIII (FVIII) inhibitors, surprisingly little is known about the in vivo mechanism of this most serious complication of replacement therapy for hemophilia A. These neutralizing antidrug alloantibodies arise in ∼30% of patients. Inhibitor formation is T-cell dependent, but events leading up to helper T-cell activation have been elusive because of, in part, the complex anatomy and cellular makeup of the spleen. Here, we show that FVIII antigen presentation to CD4+ T cells critically depends on a select set of several anatomically distinct antigen-presenting cells, whereby marginal zone B cells and marginal zone and marginal metallophilic macrophages but not red pulp macrophages (RPMFs) participate in shuttling FVIII to the white pulp in which conventional dendritic cells (DCs) prime helper T cells, which then differentiate into follicular helper T (Tfh) cells. Toll-like receptor 9 stimulation accelerated Tfh cell responses and germinal center and inhibitor formation, whereas systemic administration of FVIII alone in hemophilia A mice increased frequencies of monocyte-derived and plasmacytoid DCs. Moreover, FVIII enhanced T-cell proliferation to another protein antigen (ovalbumin), and inflammatory signaling–deficient mice were less likely to develop inhibitors, indicating that FVIII may have intrinsic immunostimulatory properties. Ovalbumin, which, unlike FVIII, is absorbed into the RPMF compartment, fails to elicit T-cell proliferative and antibody responses when administered at the same dose as FVIII. Altogether, we propose that an antigen trafficking pattern that results in efficient in vivo delivery to DCs and inflammatory signaling, shape the immunogenicity of FVIII.
Introduction
Hemophilia A is a bleeding disorder caused by mutations in the F8 gene that result in the absence or dysfunction of the coagulation factor VIII (FVIII) protein. The puzzling development of neutralizing antidrug alloantibodies (inhibitors) in response to FVIII is the most serious complication of hemophilia A replacement therapy. FVIII inhibitors arise in ∼30% of patients, thwarting the therapy.1 This is surprising given that, compared with other protein replacement therapies, a highly purified protein is administered IV at a low antigen dose. Immune tolerance induction therapy may eradicate inhibitors, but it entails burdensome FVIII treatment, and is successful only in ∼70% of individuals.2,3 Emicizumab, a bispecific antibody mimicking FVIII, has improved outcomes, but breakthrough bleeds still require additional hemostatic treatment, preferably FVIII replacement, therefore, FVIII tolerance remains desirable.4 However, the mechanism of immune response to FVIII is insufficiently understood, which has stymied development of new FVIII tolerance therapies.5,6
B-cell activation giving rise to inhibitors requires help from CD4+ T cells, which is dependent on major histocompatibility complex class II (MHC-II) presentation of FVIII peptides. Beyond this cursory understanding, little is known about the in vivo mechanisms leading to FVIII T-cell responses. Animal studies have found ICOS-ICOSL, CD28-CD86, and CD40-CD154 to be critical costimulatory pathways for antigen-presenting cell (APC)–T cell and T cell–B cell interactions.7-10 A more recent report showed that B-cell activating factor modulates FVIII inhibitor formation in humans and mice with hemophilia A.11 However, most of our knowledge on FVIII-APC interactions has come from in vitro experiments, which lack the tissue architecture of secondary lymphoid organs (SLOs), and, thus, have limited relevance for in vivo responses.12 Subsets of APCs, T cells, and B cells occupy, migrate between, or demarcate distinct compartments in the SLOs. In the spleen, which orchestrates immune responses to blood-borne antigens, this anatomic segregation may dictate responses to antigens because large molecules (>60 kDa) cannot freely flow to the T-cell zones and need to be carried across the marginal zone (MZ) populated by several types of specialized immune cells acting as gatekeepers or ferries between the red and white pulp.13 A study by Zerra et al on the role of marginal zone B cells (MZBs) in FVIII inhibitor formation first hinted that the anatomy and cellular makeup of the spleen may shape the immune response to FVIII.14
Here, we investigated the requirements for specific APCs in FVIII uptake and antigen presentation to helper T cells in vivo, the immunogenicity of FVIII compared with other T-dependent antigens, and the resulting effects on dendritic, and T and B cell responses to FVIII in mice with hemophilia A. We show that MHC-II presentation of FVIII critically depends on a select set of anatomically distinct APCs, whereby several cell types participate in shuttling the antigen to the white pulp in which dendritic cells (DCs) prime helper T cells, which differentiate into follicular helper T (Tfh) cells. Furthermore, we find that the antigen trafficking pattern and inflammatory signaling shape the immunogenicity of FVIII. In contrast to chicken ovalbumin (OVA), which fails to elicit an antibody response, FVIII is not found in red pulp macrophages (RPMFs) but instead efficiently traffics to splenic DCs.
Methods
Mouse strains and procedures
Wild-type (WT) C57BL/6, OT-II, CD11c-DTR/green fluorescent protein (GFP), and S129/C57Bl/6 hemophilia A (HA, F8e16y/–) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). MyD88–/–, IFNAR–/–, BDCA2-DTR/GFP, XCR1-DTRvenus, and C57BL/6 hemophilia A (HA, F8e16y/–) mice were bred in-house. All animals were 8- to 12-week-old males, housed and treated under Institutional Animal Care and Use Committee–approved protocols at Indiana University. Reagents were injected IV, intraperitoneally (IP), or intradermally (ID). Procedures for individual reagents and sample collection are detailed in the supplemental Material, available on the Blood website.
FVIII-AF488 and FVIII-OVA generation
Recombinant B domain deleted FVIII (FVIII-SQ)15 was donated by Bayer. The protein was labeled using 400-fold molar excess of A488 C5 maleimide (ThermoFisher Scientific, Waltham, MA) for 6 hours at 4°C in 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 0.15 M NaCl, 5 mM CaCl2, and 0.01% Tween 80, pH 7.0.16 Full reaction contents were then run over a 80-mL P-6GD column (Bio-Rad, Carlsbad, CA) and rFVIII-SQ-A488 was eluted in high-salt buffer (20 mM HEPES [N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid], 0.65 M NaCl, 5 mM CaCl2, and 0.01% Tween 80, pH 7.0).
To generate FVIII-OVA, the chicken OVA peptide consensus sequence was introduced into human FVIII-SQ15 DNA via site-directed mutagenesis by Genscript (Piscataway, NJ).17 Plasmid DNA was used to generate a baby hamster kidney cell line stably expressing FVIII-OVA, which was then purified, as previously described.18,19
Flow cytometry
Single-cell suspensions of splenocytes were prepared as published.20 Upon pretreatment with Fc block (BD Biosciences, San Diego, CA), splenocytes were stained with antibodies and a viability dye (supplemental Methods). Data were collected on LSRFortessa (BD Biosciences) and analyzed with FCS Express (De Novo Software, Glendale, CA).
FVIII inhibitor and total antibody quantification
Cell sorting and adoptive transfers
CD4+ T cells were purified from splenocytes of OT-II mice using a CD4+ T-cell isolation kit (Miltenyi Biotec, Auburn, CA) and labeled with 5 μM CellTrace Violet (CTV) (ThermoFisher Scientific). Experimental mice received 5 × 106 labeled CD4+ T cells, 1 day after antigen injection. Spleens were harvested 4 days later to measure proliferation by flow cytometry. For intravital microscopy (IVM), mice received 107 labeled CD4+ T cells, 1 day before imaging.
IVM
Inguinal lymph nodes (LNs) of anesthetized CD11c-DTR/GFP mice were surgically exposed and imaged in vivo on a Leica SP8 confocal/multiphoton microscope with a HCX IRAPO L 25x/0.95 water dipping objective (Leica Microsystems, Wetzlar, Germany). Imaging data were collected and processed using Leica LASX software.
Statistics
Results are reported as the mean ± standard deviation (SD) or median ± interquartile ranges (IQRs). Significant differences were determined with unpaired 2-tailed Student t test or one-way analysis of variance with Tukey posttests. P < .05 were considered significant (∗P < .05; ∗∗P < .01; ∗∗∗P < .001; and ∗∗∗∗P < .0001). Analyses were performed using GraphPad Prism (San Diego, CA).
Results
Multiple types of APCs take up FVIII upon systemic administration
To determine which APCs capture FVIII in vivo, we labeled FVIII with Alexa Fluor (AF) 488 and administered the labeled protein to S129/C57Bl/6 HA mice (Figure 1A). Results were contrasted with those from animals receiving ovalbumin (OVA)-AF488. After 30 to 180 minutes, either protein was detectable in multiple types of DCs, macrophages (MFs), and B cells in the spleen, but protein distribution across the analyzed cell types differed substantially (Figure 1B-C; supplemental Figure 2A). FVIII-AF488 preferentially localized in conventional DC type 1 cells (cDC1s), with the percentage of FVIII-AF488+ cDC1s being 5-fold higher than FVIII-AF488+ conventional DC type 2 cells (cDC2s) (P < .0001). RPMFs had little or no detectable FVIII-AF488. In contrast, in animals that received OVA-AF488, RPMFs internalized the most antigen, with 29-fold and 26-fold more OVA-AF488+ RPMFs than cDC1s and cDC2s, respectively (P < .0001), at the lower dose of OVA-AF488 (Figure 1B). Neither protein was detectable in plasmacytoid DCs (pDCs), except OVA-AF488 at the higher dose in some animals. Percentages of FVIII-AF488+ APCs peaked 60 minutes after administration, declining at 180 minutes (Figure 1C). OVA-AF488+ APCs revealed a similar pattern, except cDC1s and MZ macrophages (MZMs) peaked at 30 minutes and then declined at later time points.

RPMFs robustly take up OVA (a “weak” immunogen) but not FVIII. (A) Experimental timeline. S129/C57BL/6 HA mice were injected IV with FVIII-AF488 or OVA-AF488 (n = 3-5 per group). After 30 to 180 minutes, the spleens were collected for flow cytometry. (B) Quantification of FVIII-AF488+ or OVA-AF488+ cells in cDC1s (CD8α+CD11b–CD11c+), cDC2s (CD8α–CD11b+CD11c+), pDCs (PDCA-1+CD11c+), and RPMFs (F4/80+CD11b–) 1 hour after administration of 5 μg FVIII-AF488 or OVA-AF488. (C) Quantification of FVIII-AF488+ or OVA-AF488+ cells in cDC1s (CD8α+CD11b–CD11c+), cDC2s (CD8α–CD11b+CD11c+), pDCs (PDCA-1+CD11c+), RPMFs (F4/80+CD11b–), MZMs (F4/80loCD11b+TIM-4hi), MMMs (F4/80loCD11b+TIM-4lo), MZ B cells (B220+CD21hiCD23lo/–), transitional B cells (B220+CD21−CD23lo/−), and follicular B cells (B220+CD21medCD23lo/–), 30 to 180 minutes after injection of 10 μg FVIII-AF488 or OVA-AF488. Shown are means ± SD, and P values from analysis of variance. MMMS, marginal metallophilic macrophages.
RPMFs robustly take up OVA (a “weak” immunogen) but not FVIII. (A) Experimental timeline. S129/C57BL/6 HA mice were injected IV with FVIII-AF488 or OVA-AF488 (n = 3-5 per group). After 30 to 180 minutes, the spleens were collected for flow cytometry. (B) Quantification of FVIII-AF488+ or OVA-AF488+ cells in cDC1s (CD8α+CD11b–CD11c+), cDC2s (CD8α–CD11b+CD11c+), pDCs (PDCA-1+CD11c+), and RPMFs (F4/80+CD11b–) 1 hour after administration of 5 μg FVIII-AF488 or OVA-AF488. (C) Quantification of FVIII-AF488+ or OVA-AF488+ cells in cDC1s (CD8α+CD11b–CD11c+), cDC2s (CD8α–CD11b+CD11c+), pDCs (PDCA-1+CD11c+), RPMFs (F4/80+CD11b–), MZMs (F4/80loCD11b+TIM-4hi), MMMs (F4/80loCD11b+TIM-4lo), MZ B cells (B220+CD21hiCD23lo/–), transitional B cells (B220+CD21−CD23lo/−), and follicular B cells (B220+CD21medCD23lo/–), 30 to 180 minutes after injection of 10 μg FVIII-AF488 or OVA-AF488. Shown are means ± SD, and P values from analysis of variance. MMMS, marginal metallophilic macrophages.
Systemic administration of FVIII alters DC numbers in the spleen
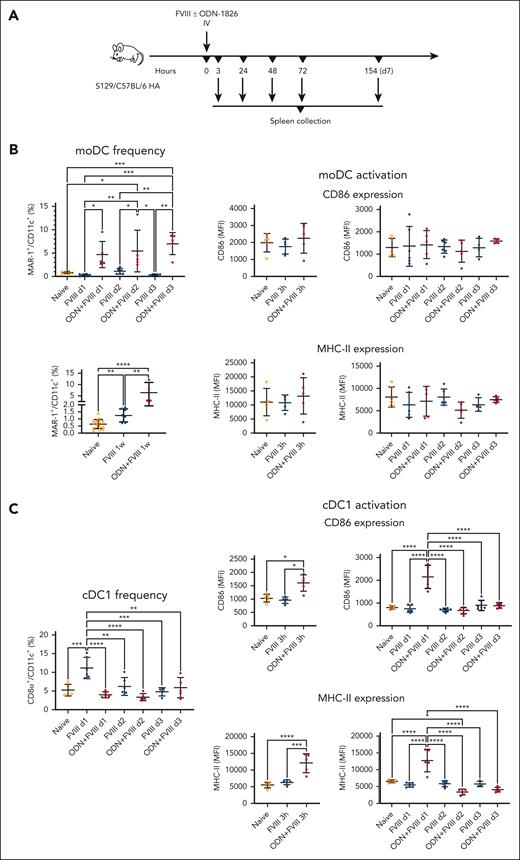
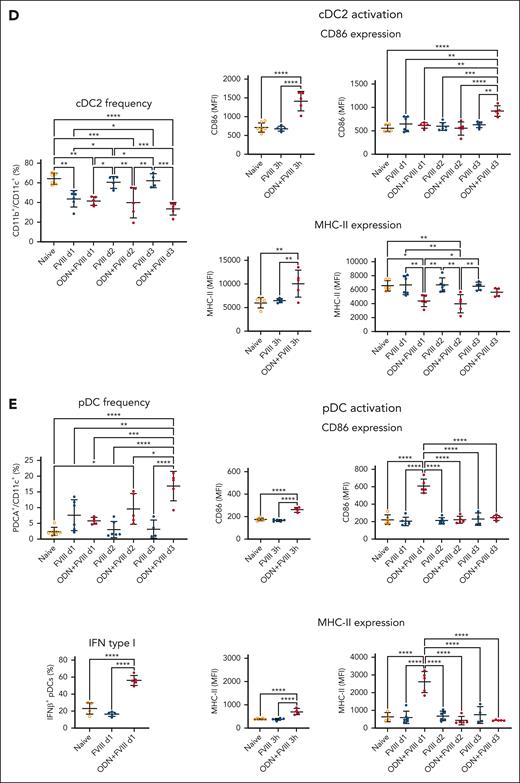
We also asked the question whether a single clinical dose of FVIII could produce discernible changes in splenic DC numbers and activation status. To that end, we injected FVIII (1.5 IU) alone or in combination with a potent activator of innate immunity, ODN-1826, into S129/C57Bl/6 HA mice (Figure 2A). This Toll-like receptor 9 (TLR9) agonist enhances antibody formation by increasing Tfh cell responses via monocyte-derived DCs (moDCs), thereby allowing us to assess responses depending on the presence of a well-characterized FVIII-extrinsic innate activation or “danger” signal.23-26 One week after administration, moDC frequencies were ∼2.5-fold higher in the animals that received only FVIII. In the coinjected group (FVIII + ODN-1826), moDCs were increased at all time points (3 hours; 1, 2, and 3 days; and 1 week after a single injection) with the peak (∼10-fold increase) reached at 72 hours and remaining similarly elevated after 1 week (Figure 2B). Conversely, cDC1 frequencies trended higher at day 1 to day 3 after FVIII injection (∼2.1 fold at day 1), whereas the coinjected animals had mostly unchanged cDC1 frequencies. Moreover, this subset showed >1.5-fold upregulation of CD86 and MHC-II in the coinjected group, 3 to 24 hours after injection (Figure 2C). In contrast, we found 1.5-fold lower frequencies of cDC2s at day 1 in both groups (FVIII with and without ODN1826), with the coinjected animals having similarly decreased cDC2 frequencies between days 2 and 3. In the coinjected mice, CD86 and MHC-II were elevated ∼2 fold at 3 hours but then MHC-II declined and remained significantly downregulated between day 1 and day 2 (Figure 2D). Furthermore, we found significantly increased frequencies of pDCs in the coinjected group at day 2 and day 3 after injection (∼4- and ∼18-fold increase, respectively). In mice injected only with FVIII, pDC frequencies peaked at day 1 (elevated ∼3 fold). This subset showed significantly upregulated CD86 and MHC-II in the coinjected group at 3 to 24 hours after injection, which also had >2-fold higher levels of interferon β (IFN-β) at 24 hours (Figure 2E). The total cell numbers also markedly changed in response to FVIII with or without ODN1826 for all subsets except pDCs, with changes in the number of moDC and cDC2 mirroring changes in frequency. These frequency and number variations were distinct from the effects of ODN1826 alone, showing that the changes observed in response to coinjections were the combined effects of FVIII and ODN (supplemental Figures 3-4).
Systemic administration of FVIII with or without TLR9 agonist alters DC numbers in the spleen. (A) Experimental timeline. S129/C57BL/6 HA mice received a single injection of FVIII (1.5 IU) +/– ODN1826 (50 μg) IV (n = 4-16 per group). After 3 to 154 hours (3 hours, or 1, 2, 3, and 7 days) after administration, the spleens were collected for flow cytometry analysis. (B) Flow cytometry analysis of moDC (MAR-1+CD64+CD11b+CD11c+) frequencies (1, 2, and 3 days, and 1 week after administration) and expression of activation markers CD86 and MHC-II on moDCs (3 hours or 1, 2, and 3 days after administration). (C) Flow cytometry analysis of cDC1 (CD8α+ CD11b–CD11c+) frequencies (1, 2, and 3 days after administration) and expression of activation markers CD86 and MHC-II on cDC1s (3 hours, or 1, 2, and 3 days after administration). (D) Flow cytometry analysis of cDC2 (CD8α–CD11b+CD11c+) frequencies (1, 2, and 3 days after administration) and expression of activation markers CD86 and MHC-II on cDC2s (3 hours, and 1, 2, and 3 days after administration). (E) Flow cytometry analysis of pDC (PDCA-1+CD11c+) frequencies (1, 2, and 3 days after administration), expression of activation markers CD86 and MHC-II on pDCs (3 hours, and 1, 2, and 3 days after administration), and intracellular IFN-β level in pDCs (1 day after administration). All plots (B-E) show means ± SD and P values by analysis of variance.
In vivo CD4+ T-cell proliferative response to FVIII requires several types of APCs
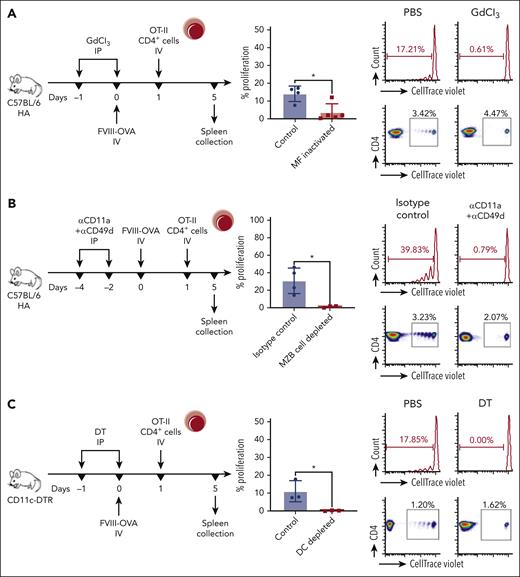
To determine which APCs are required for the CD4+ T-cell response to FVIII, we generated a recombinant FVIII protein (FOVA) that contains the peptide sequence ISQAVHAAHAEINEAGR (the immunodominant CD4+ T-cell epitope of the model antigen OVA) in place of the B domain, and that shows a similar specific activity and induces similar FVIII inhibitor responses to B domain–deleted FVIII (Figure 3A-B). Next, we used FOVA to test requirements for several APCs in a highly sensitive in vivo assay based on proliferation of adoptively transferred transgenic OT-II CD4+ T cells. OT-II cells proliferated upon transfer into C57BL/6 mice that had received FOVA but not in mice that had received FIX (an irrelevant antigen). Interestingly, CD4+ T cells required 10-fold more OVA antigen than FOVA to proliferate. Coinjection of FVIII with full-length OVA also enhanced proliferation (Figure 3C). We assayed OT-II CD4+ cell proliferation in response to FOVA in several transgenic mouse strains upon depletion of APCs populating different splenic compartments using previously described depletion protocols.14,27-29 Macrophage (MF) inactivation using gadolinium chloride abolished T-cell proliferation in all but 1 hemophilia A mouse (P < .05) (Figure 4A). Next, we depleted MZBs, which abrogated proliferation in all mice (P < .05) (Figure 4B).

Systemically administered FVIII-OVA induces more robust in vivo proliferation of CD4+ T cells than OVA. (A) Schematic diagram of FVIII-OVA (FOVA) in comparison with WT FVIII. In FOVA, the B domain was replaced with the immunodominant MHC-II epitope of chicken OVA. (B) Sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis of and inhibitor formation in response to FOVA (mean ± SD). Sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis of 2 μg rFVIII-SQ and rFVIII-SQ-OVA in the presence or absence of 100 nM thrombin. The gel was stained with Coomassie blue. A comparison with a commercially available rFVIII, ReFacto, is shown. Specific activity was determined by partial thromboplastin time one-stage clotting assay measurement relative to protein concentration. Data are shown as mean ± standard error measurements from duplicate experiments. For inhibitor assay, C57BL/6 HA mice received 4 weekly IV injections of 1.5 IU (300 ng) FOVA or B domain-deleted (BDD)-FVIII. Inhibitor titers were measured 1 week after the last injection. (C) WT C57BL/6 mice (n = 3-6) received 5 μg FOVA, full-length OVA (5 μg or 50 μg), BDD-FVIII (5 μg) + full-length OVA (50 μg), or FIX (5 μg) IV. One day later, CD4+ T cells were magnetically sorted from OT-II mice, labeled with CTV, and adoptively transferred to the experimental C57BL/6 mice. Four days later the spleens were collected for flow cytometry analysis of proliferation. The plots show a summary of percentage proliferation (mean ± SD), representative proliferation histograms, and respective dot plots.
Systemically administered FVIII-OVA induces more robust in vivo proliferation of CD4+ T cells than OVA. (A) Schematic diagram of FVIII-OVA (FOVA) in comparison with WT FVIII. In FOVA, the B domain was replaced with the immunodominant MHC-II epitope of chicken OVA. (B) Sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis of and inhibitor formation in response to FOVA (mean ± SD). Sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis of 2 μg rFVIII-SQ and rFVIII-SQ-OVA in the presence or absence of 100 nM thrombin. The gel was stained with Coomassie blue. A comparison with a commercially available rFVIII, ReFacto, is shown. Specific activity was determined by partial thromboplastin time one-stage clotting assay measurement relative to protein concentration. Data are shown as mean ± standard error measurements from duplicate experiments. For inhibitor assay, C57BL/6 HA mice received 4 weekly IV injections of 1.5 IU (300 ng) FOVA or B domain-deleted (BDD)-FVIII. Inhibitor titers were measured 1 week after the last injection. (C) WT C57BL/6 mice (n = 3-6) received 5 μg FOVA, full-length OVA (5 μg or 50 μg), BDD-FVIII (5 μg) + full-length OVA (50 μg), or FIX (5 μg) IV. One day later, CD4+ T cells were magnetically sorted from OT-II mice, labeled with CTV, and adoptively transferred to the experimental C57BL/6 mice. Four days later the spleens were collected for flow cytometry analysis of proliferation. The plots show a summary of percentage proliferation (mean ± SD), representative proliferation histograms, and respective dot plots.
CD4+ T-cell proliferation in response to FVIII requires a select set of APCs. (A) Inactivation of MFs. C57BL/6 HA mice (n = 4-5) received GdCl3 or PBS (control) IP, 1 day before, and on, the day of IV FOVA (5 μg) administration. (B) Depletion of MZ B cells. C57BL/6 HA mice (n = 3-4) received anti-CD11a and anti-CD49d or isotype control antibodies IP, 4 and 2 days before IV FOVA (5 μg) administration. (C) Depletion of DCs, MZMs, and MMMs. C57BL/6 CD11c-DTR/GFP mice (n = 3) received DT or PBS (control) IP, 1 day before, and on, the day of IV FOVA (5 μg) administration. (D) Depletion of pDCs. C57BL/6 BDCA2-DTR/GFP mice (n = 5) received DT or PBS (control) IP 1 day before, 1 day after, and 3 days after IV FOVA (5 μg) administration. (E) Depletion of cDC1s. C57BL/6 XCR1-DTRvenus mice (n = 4) received DT or PBS (control) IP, 1 day before and 2 days after IV FOVA (5 μg) administration. (F) S129/C57BL/6 HA mice (n = 5-12 per group) received a single FVIII +/− ODN1826 injection after MZ B-cell depletion. One week later, pDC frequencies were analyzed by flow cytometry in naïve (control), FVIII control (FVIII + isotype), ODN + FVIII control (ODN + FVIII + isotype), and MZ B cell–depleted (FVIII + αMZB and ODN + FVIII + αMZB) mice. Shown are means ± SD and P values by analysis of variance. (A-E) In each experiment, 1 day after FOVA administration, CD4+ T cells were magnetically sorted from OT-II mice, labeled with CTV and adoptively transferred to the experimental animals. Four days later the spleens were collected for flow cytometry analysis of proliferation. The plots show a summary of percentage proliferation (mean ± SD), representative proliferation histograms, and respective dot plots. P values come from unpaired Student t test. GdCl3, gadolinium chloride; MMMS, marginal metallophilic macrophages; PBS, phosphate buffered saline.
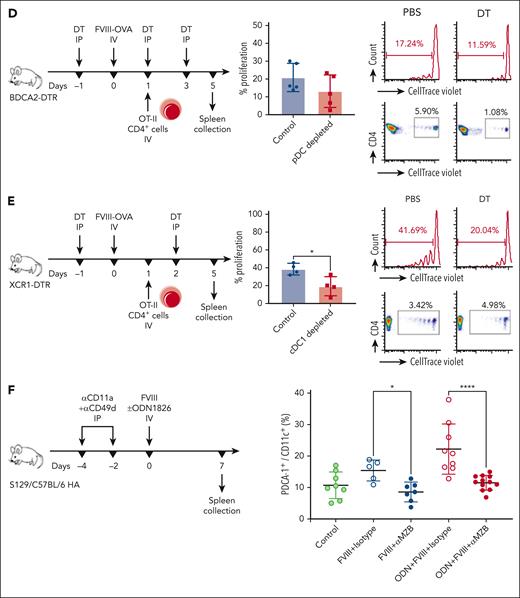
We also used conditional knockout mice expressing diphtheria toxin receptor (DTR) in different APC subsets, which allowed for targeted depletion using diphtheria toxin (DT).27-29 Depletion of DCs in CD11c-DTR/GFP mice abrogated T-cell proliferation (P < .05) (Figure 4C). Although DT treatment depletes most DCs in CD11c-DTR/GFP mice (including cDC1 and cDC2) as well as MZMs and marginal metallophilic macrophages, it does not deplete pDCs, because of their lower expression of CD11c. Therefore, the complete abrogation of proliferation suggested that pDCs do not present FOVA peptides, consistent with the FVIII-AF488 uptake results. Similarly, animals depleted of pDCs showed only slightly, nonsignificantly reduced proliferation (Figure 4D). To define the role of cDC1s, which were the main subset that took up FVIII-AF488, we used XCR1-DTRvenus mice. Depletion of XCR1+ cells (80% of which are cDC1s) in these animals reduced T-cell proliferation by ∼50% (P < .05) (Figure 4E), indicating that cDC1s contribute to FVIII peptide presentation to helper T cells. Collectively, these results showed that multiple APC subsets are required and likely cooperate in priming helper T-cell responses to FVIII.
Cognate interaction with APCs drives CD4+ T cells to the T-B border
To track and visualize early interactions between immune cells in vivo after FOVA administration, we performed multiphoton IVM of inguinal LNs in CD11c-DTR/GFP mice. Because the microanatomy of LNs closely resembles the white pulp and because skin-draining LNs are more amenable to IVM than the spleen, we drove immune responses to inguinal LNs by injecting FOVA intradermally in the groin area of mice that received labeled OT-II CD4+ cells on the previous day. CTV+ cells were nearly absent from LNs in animals that did not receive FOVA, and in animals 1 hour after FOVA injection. GFP+ cells were visible across LNs and, together with the second harmonic generation signals (coming from collagen), clearly demarcated the LN borders and B-cell follicles (Figure 5A; supplemental Movie 1). Five hours after FOVA injection, multiple CD4+CTV+ cells formed clusters around GFP+ cells throughout the T-cell zone (Figure 5B), with several motile CD4+CTV+ cells surveying the B-cell follicle (supplemental Movie 2). Twenty hours after FOVA injection, CD4+CTV+ cells crowded and demarcated the T cell–B cell (T-B) border, suggesting their differentiation into Tfh cells (Figure 5C; supplemental Movie 3). LNs of animals that had received FIX (an irrelevant antigen) 20 hours before microscopy were more populated by CTV+ cells compared with naïve animals, but they appeared randomly distributed across the tissue (supplemental Figure 6).

Interaction with APCs in response to FVIII drives CD4+ T cells to the T-B border. (A) Multiphoton IVM of inguinal LNs in C57BL/6 CD11c-DTR/GFP mice. Twenty-four hours before microscopy, CD4+ T cells were magnetically sorted from OT-II mice, labeled with CTV, and adoptively transferred to the experimental animals. FOVA (5 μg) was injected intradermally in the groin area 1, 5, or 20 hours (n = 2 per time point) before microscopy. LNs were surgically exposed in anesthetized mice and imaged in vivo on an inverted Leica SP8 confocal/multiphoton microscope with a HCX IRAPO L 25×/0.95 water dipping objective. During imaging, the core body temperature of the animals was maintained at 36°C to 37°C with a temperature controller consisting of a rectal probe and a heating pad. Imaging data were collected and processed using Leica LASX software. Sixty-minute time series were captured with Z-stacks collected once every minute for each time point to a depth of ∼100 μm. The full-field views show whole LNs in animals without a FOVA injection (no antigen) and 1 hour after a FOVA (5 μg) injection, with observable CD11c+GFP+ cells (green), collagen (gray), and CD4+CTV+ cells (purple). The schematic diagram indicates presumed LN compartments with areas densely populated by bright CD11c+GFP+ cells (green) representing the T-cell zones and dark round areas representing the B-cell follicles. (B) Optical section and Z-stack projection views of T-cell zone and B-cell follicle areas, 5 hours after a FOVA (5 μg) injection. White triangles indicate an example CD4+ T-cell cluster in the T-cell zone around a CD11c+GFP+ cell interacting with 2 different T cells, 2 minutes apart. (C) Optical section of the T-B border 20 hours after a FOVA (5 μg) injection.
Interaction with APCs in response to FVIII drives CD4+ T cells to the T-B border. (A) Multiphoton IVM of inguinal LNs in C57BL/6 CD11c-DTR/GFP mice. Twenty-four hours before microscopy, CD4+ T cells were magnetically sorted from OT-II mice, labeled with CTV, and adoptively transferred to the experimental animals. FOVA (5 μg) was injected intradermally in the groin area 1, 5, or 20 hours (n = 2 per time point) before microscopy. LNs were surgically exposed in anesthetized mice and imaged in vivo on an inverted Leica SP8 confocal/multiphoton microscope with a HCX IRAPO L 25×/0.95 water dipping objective. During imaging, the core body temperature of the animals was maintained at 36°C to 37°C with a temperature controller consisting of a rectal probe and a heating pad. Imaging data were collected and processed using Leica LASX software. Sixty-minute time series were captured with Z-stacks collected once every minute for each time point to a depth of ∼100 μm. The full-field views show whole LNs in animals without a FOVA injection (no antigen) and 1 hour after a FOVA (5 μg) injection, with observable CD11c+GFP+ cells (green), collagen (gray), and CD4+CTV+ cells (purple). The schematic diagram indicates presumed LN compartments with areas densely populated by bright CD11c+GFP+ cells (green) representing the T-cell zones and dark round areas representing the B-cell follicles. (B) Optical section and Z-stack projection views of T-cell zone and B-cell follicle areas, 5 hours after a FOVA (5 μg) injection. White triangles indicate an example CD4+ T-cell cluster in the T-cell zone around a CD11c+GFP+ cell interacting with 2 different T cells, 2 minutes apart. (C) Optical section of the T-B border 20 hours after a FOVA (5 μg) injection.
TLR9 stimulation accelerates follicular helper T cell, GC B cell, and inhibitor responses to FVIII
In an attempt to link the observations that CD4+ T cells localized to the T-B border in response to FVIII, and that TLR9 stimulation enhances early DC responses, we assessed whether a clinically relevant FVIII dose (1.5 IU) could induce Tfh cell responses along with germinal center (GC) and inhibitor formation in hemophilia A mice, and whether TLR9 stimulation could affect these responses. After 4, 6, and 8 weekly injections, we found increased numbers of Tfh cells in the spleen, both in mice injected only with FVIII and coinjected mice (FVIII with ODN-1826), with the frequencies being 1.4-, 12.2- and 1.65-fold higher, respectively, in the coinjected animals [F(8121) = 12.32; P < .0001] (Figure 6A-B). Splenic GC B-cell numbers were similarly increased in both groups, and 2.9-, 5.3-, and 1.1-fold higher in the coinjected animals than in the FVIII-only group after 4, 6, and 8 weekly injections, respectively [F(8118) = 8.975; P < .0001]. In the FVIII-only group, both Tfh and GC B cells contracted after 6 weekly injections, likely signifying further differentiation into memory T cells and plasma cells, respectively, and their exit from the spleen. Both Tfh and GC B cells increased again after 8 injections. In contrast, the coinjected animals had peak Tfh and GC B-cell numbers after 6 injections, which declined after 8 injections, suggesting altered dynamics of differentiation and trafficking of these cells under TLR9 stimulation.

TLR9 stimulation accelerates Tfh cell, GC B cell, and antibody responses to FVIII. (A) Experimental timeline. S129/C57BL/6 HA mice received repeated (4, 6, or 8) weekly injections of FVIII (1.5 IU) with or without ODN1826 (50 μg) IV (n = 5-27 per group). Blood samples and the spleens were collected for flow cytometry analysis, ELISA, and Bethesda assays, 1 week after the last injection. (B) Tfh cell (CD4+CXCR5+PD-1+Bcl-6+FoxP3−) and GC B cell (CD19+CD95+GL7+) frequencies after 4, 6, or 8 weekly injections of FVIII (1.5 IU) with or without ODN1826 (50 μg) IV. (C) Inhibitor and FVIII-specific IgG1 titers after 4, 6, or 8 weekly injections of FVIII (1.5 IU) with or without ODN1826 (50 μg) IV. (D) Correlations of Tfh or GCB cell frequencies and inhibitor titers. P values and 95% CIs were determined for Pearson correlation coefficients. (E) Inhibitor titers (mean ± SD) in S129/C57BL/6 HA mice that received 4 weekly injections of FVIII only or FVIII and ODN1826 administered separately on 2 consecutive days (first ODN1826, then FVIII on the following day). (B and C) Shown are mean ± SD and P values from analysis of variance. ELISA, enzyme-linked immunosorbent assay.
TLR9 stimulation accelerates Tfh cell, GC B cell, and antibody responses to FVIII. (A) Experimental timeline. S129/C57BL/6 HA mice received repeated (4, 6, or 8) weekly injections of FVIII (1.5 IU) with or without ODN1826 (50 μg) IV (n = 5-27 per group). Blood samples and the spleens were collected for flow cytometry analysis, ELISA, and Bethesda assays, 1 week after the last injection. (B) Tfh cell (CD4+CXCR5+PD-1+Bcl-6+FoxP3−) and GC B cell (CD19+CD95+GL7+) frequencies after 4, 6, or 8 weekly injections of FVIII (1.5 IU) with or without ODN1826 (50 μg) IV. (C) Inhibitor and FVIII-specific IgG1 titers after 4, 6, or 8 weekly injections of FVIII (1.5 IU) with or without ODN1826 (50 μg) IV. (D) Correlations of Tfh or GCB cell frequencies and inhibitor titers. P values and 95% CIs were determined for Pearson correlation coefficients. (E) Inhibitor titers (mean ± SD) in S129/C57BL/6 HA mice that received 4 weekly injections of FVIII only or FVIII and ODN1826 administered separately on 2 consecutive days (first ODN1826, then FVIII on the following day). (B and C) Shown are mean ± SD and P values from analysis of variance. ELISA, enzyme-linked immunosorbent assay.
TLR9 stimulation robustly enhanced inhibitor formation, producing ∼8-fold higher inhibitor titers in the coinjected mice than in the FVIII-only group after 4 weekly injections (Figure 6C). Additional coinjections did not significantly increase inhibitor titers, in contrast to continued FVIII dosing, which produced higher titers after 6 and 8 weekly injections, with the 8-week titers being similar between the FVIII-only and the coinjected animals. Total anti-FVIII IgG1 antibody levels followed a similar pattern, with differences between the FVIII-only and the coinjected mice narrowing upon continued injections between 4 and 8 weeks after initial doses. This suggests that TLR9 stimulation accelerates inhibitor development, which thus reaches peak magnitudes earlier. Both Tfh and GC B-cell numbers correlated with inhibitor titers ([r(31) = 0.51, P = .0025; and r(31) = 0.52; P = .002, respectively]; Figure 6D).
Induction of antibody responses to FVIII is reduced without MyD88 signaling
Next, we asked whether inflammatory signaling pathways play a role in FVIII inhibitor responses without additional stimulation. Because TLRs and interleukin-1 receptor send signals through the universal adapter protein myeloid differentiation factor 88 (MyD88), we used C57BL/6 MyD88−/− mice to evaluate inhibitor responses in the absence of inflammatory signaling pathways. In addition, we assessed inhibitor formation in the absence of type I IFN signaling using C57BL/6–type I IFN receptor (IFNAR)–/– mice because IFNAR relays signals independently from MyD88. After 4 weekly injections, average inhibitor and total anti-FVIII IgG1 titers were not significantly different between the control and signaling-deficient mice, but the MyD88–/– group had a markedly larger number of nonresponders (Figure 7A-B). The odds of MyD88–/– mice developing FVIII inhibitors were 50 fold lower (odds ratio [OR] = 0.13; 95% confidence interval [CI], 0.017-0.71) compared with the control animals (OR, 6.5; 95% CI, 1.8-42). Animals that did mount antibody responses had similar inhibitor and anti-FVIII IgG1 titers to those of controls, suggesting that MyD88 signaling–deficient animals can develop unimpeded inhibitor responses but are more likely to tolerate FVIII. Although IFNAR–/– mice also seemed less likely to develop FVIII inhibitors, they had similar anti-FVIII IgG1 responses, suggesting that type I IFN signaling does not affect the tolerance threshold but it may enhance neutralizing antibody responses to FVIII. The effect of type I IFN may be limited to B-cell activation because IFNAR–/– mice, surprisingly, exhibited elevated helper T-cell proliferation to FOVA in our in vivo antigen presentation assay (supplemental Figure 5).

Inflammatory signaling–deficient mice are less likely to develop FVIII inhibitors. (A) Experimental timeline. WT C57BL/6, MyD88–/–, and IFNAR–/– mice received 4 weekly injections of FVIII (2.5 IU) IV (n = 13-15 per group). Blood samples were collected for ELISA and Bethesda assays 1 week after the last injection. (B) Inhibitor and FVIII-specific IgG1 titers in WT and knockout mice 1 week after the last FVIII injection. The dotted horizontal line is an approximate sensitivity cut-off for ELISA. Shown are medians ± IQRs. (C) Experimental timeline. S129/C57BL/6 HA mice received 4 weekly injections of FVIII, FIX, or OVA (300 ng each) IV (n = 7-9 per group). Blood samples were collected for ELISA and Bethesda assays 1 week after the last injection. (D) FVIII-, FIX-, or OVA-specific IgG1 titers 1 week after the last injection. Shown are mean ± SD, and P values from analysis of variance. ELISA, enzyme-linked immunosorbent assay.
Inflammatory signaling–deficient mice are less likely to develop FVIII inhibitors. (A) Experimental timeline. WT C57BL/6, MyD88–/–, and IFNAR–/– mice received 4 weekly injections of FVIII (2.5 IU) IV (n = 13-15 per group). Blood samples were collected for ELISA and Bethesda assays 1 week after the last injection. (B) Inhibitor and FVIII-specific IgG1 titers in WT and knockout mice 1 week after the last FVIII injection. The dotted horizontal line is an approximate sensitivity cut-off for ELISA. Shown are medians ± IQRs. (C) Experimental timeline. S129/C57BL/6 HA mice received 4 weekly injections of FVIII, FIX, or OVA (300 ng each) IV (n = 7-9 per group). Blood samples were collected for ELISA and Bethesda assays 1 week after the last injection. (D) FVIII-, FIX-, or OVA-specific IgG1 titers 1 week after the last injection. Shown are mean ± SD, and P values from analysis of variance. ELISA, enzyme-linked immunosorbent assay.
We also compared the immunogenicity of FVIII with 2 other T-dependent antigens, FIX and OVA, by repeat dosing of each protein (300 ng) to S129/C57Bl/6 HA mice. After 4 weekly injections, only the FVIII-injected mice developed IgG1 antibodies in response to the antigen (Figure 7C-D; supplemental Figure 7), supporting that FVIII has intrinsic immunostimulatory properties.
Discussion
Delivery and MHC-II presentation of the FVIII antigen to CD4+ T cells requires a concerted action of MFs, MZ B cells, and DCs
Despite >80 years of clinical experience with FVIII inhibitors, surprisingly little is known about the in vivo mechanism of this most serious complication of FVIII replacement therapy.5,30,31 The cellular underpinnings of T-cell response to FVIII have been elusive. MHC-II presentation requires that the FVIII antigen reaches the SLOs (primarily the spleen), in which it needs to be captured, moved to the relevant location, processed, and loaded onto MHC-II for presentation to the cognate-naïve CD4+ T cells. To complete this journey, FVIII has to traverse several tissue compartments populated by distinct immune cells.13 In vitro studies cannot recapitulate this complex mise-en-scène. Previous reports highlighted the role of individual APC types in the initiation of adaptive responses to FVIII.14,32-34 Here, we find that upon systemic administration, multiple types of splenic MFs, B cells, and DCs take up FVIII in vivo, and that several DC types increase and/or are activated in the first hours and days after administration of a clinically relevant FVIII dose. To interrogate the roles of these different APCs and integrate observations from previous studies, we developed an in vivo antigen presentation assay, which ensured that MHC-II presentation followed the sequence of interactions that occur in the setting of FVIII replacement therapy. We find that in vivo priming of CD4+ T cells in response to FVIII cannot proceed in the concomitant absence of MZMs, marginal metallophilic macrophages, and conventional DCs, or select absence of MFs or MZ B cells. Select depletion of cDC1s partially reduced helper T-cell priming.
Among DCs, cDC1s took up the most FVIII and represent a major DC subset that presents FVIII to CD4+ T cells. Although cDC1s have a specialized function in crosspresentation of exogenous antigens on MHC-I to CD8+ T cells, they are also important MHC-II presenters, which, for example, allows them to serve as a platform for CD4+ and CD8+ cell interactions in antiviral responses.35 In contrast, pDCs are not directly involved in FVIII antigen presentation but likely provide activation signals, given their increase in frequency and known innate functions. MFs are not likely to directly present FVIII because they remain in the MZ, whereas in IVM we saw CD4+ T-cell clustering deep in the T-cell zone, away from the fringes. In contrast, MZ B cells do travel to the white pulp but they did not rescue T-cell proliferation in the concomitant absence of MFs and DCs, therefore MZ B cells are also unlikely to present. Conversely, in the absence of MZ B cells, helper T-cell proliferation was abrogated and pDC frequencies did not rise in response to FVIII, even when coinjected with an adjuvant (Figure 4F), suggesting that FVIII did not reach the white pulp. These observations and the arrangement of splenic APCs between the red and white pulp support that they participate in trafficking of FVIII to T cells, perhaps in a bucket relay–like manner. APCs can share antigen through mechanisms independent of antigen-receptor recognition, such as trogocytosis.36-38
Conventional DC2s are tissue-resident DCs that predominate in the spleen, in contrast to moDCs, which differentiate in situ from monocytes in response to inflammatory signals.39 Increases in moDC frequencies that we detected at 1 week in response to FVIII were delayed in relation to changes in cDC1, cDC2, and pDC numbers as well as relocation of CD4+ T cells (all of which occurred within hours or days after administration), except when FVIII was coinjected with a TLR9 agonist, which triggered a rapid rise in moDC numbers within 24 hours. Both cDC2s and moDCs can induce Tfh cell responses and perhaps either can prime CD4+ T cells against FVIII, depending on the inflammatory milieu.
Altogether, in vivo priming of CD4+ T cells in response to FVIII requires a concerted action of multiple types of APCs that are in distinct lymphoid tissue compartments that FVIII traverses en route to cognate helper T cells. We propose that select MFs and MZ B cells shuttle the antigen to DCs, which are the final conduit for FVIII on its journey from the circulation to CD4+ T cells (supplemental Figure 8).
TLR9 stimulation accelerates inhibitor formation through enhanced Tfh cell responses
The traditional view that B cells receive help from T helper–cell type-2 cells during humoral responses has made way for an updated model with Tfh cells driving GC and antibody development. Evidence that Tfh cells induce FVIII inhibitor responses has also begun to emerge.40 Unlike non-Tfh effector T cells, upon priming by DCs, Tfh cells stay in lymphoid organs and migrate to the T-B border to provide help to B cells, thus initiating GC formation.41 Here, we found that within 24 hours after FVIII administration, CD4+ T cells migrate to, and crowd the T-B border, consistent with Tfh cell behavior, with some entering and surveying B-cell follicle. We further found that clinically relevant FVIII doses trigger elevations in the number of Tfh and GC B cells in the spleen, correlating with inhibitor formation, which is robustly accelerated by TLR9 stimulation.
TLR9 is an innate immune sensor of unmethylated CpG sequences in DNA, which are a pathogen-associated molecular pattern. Many studies explored the role of danger signals in inhibitor formation, such as vaccination, infections (generating pathogen-associated molecular patterns), or tissue damage.42 Presumably, these signals may activate innate immune sensors, such as TLRs, and thus inadvertently trigger adaptive immune responses to FVIII. Indeed, bacterial lipopolysaccharide, which is a TLR4 ligand, enhances FVIII inhibitor formation in hemophilic mice.43 Although human and murine immune cells differ in expression of TLRs, RNA sequencing analyses have revealed upregulation of several genes involved in innate immune activation and inflammation in patients with inhibitors, including TLR8 and NLRP3.44 Previous reports from our team and others have shown increased antibody responses by TLR9 stimulation through activation of moDCs, which led to enhanced Tfh cell responses.23-26 Those findings prompted us to explore the effects of TLR9 stimulation on FVIII inhibitor development. The hypothesis that innate signals may increase inhibitor risk has raised concerns over concurrent administration of vaccines and FVIII.42 However, the role of vaccination was challenged when influenza vaccine surprisingly reduced anti-FVIII responses in mice with hemophilia A.45 The largest observational study involving 375 previously untreated patients with hemophilia A also failed to find increased risk.46 Notably, most current vaccines do not contain TLR9 agonists, but many formulations with TLR9-targeting adjuvants are being evaluated in clinical trials, which will behoove caution in vaccinating people with hemophilia in the future.47 Spacing injections of such vaccines and FVIII could minimize the potential risk because we did not find increased inhibitor formation when a TLR9 agonist and FVIII were administered on separate days (Figure 6E).
Efficient trafficking of FVIII to site of T-cell activation and evidence for intrinsic immunogenicity
Several studies investigated the impact of FVIII structure and function on its immunogenicity, often with conflicting results.48-51 Here, we found that FVIII increased frequencies of DC subsets with innate/inflammatory immune functions (moDCs and pDCs) and was able to enhance T-cell proliferation to an unrelated protein antigen without a need for an adjuvant. Furthermore, mice with disrupted inflammatory signaling were less likely to develop FVIII inhibitors. Together, these findings suggest that FVIII has intrinsic immunostimulatory properties, which affect the decision of whether to mount a response. In the clinic, FVIII inhibitors occur ∼5-fold more often in hemophilia A than FIX inhibitors do in hemophilia B, despite ∼17-fold higher FIX protein doses.52 We found that FIX and OVA fail to elicit antibodies at protein doses equal to a clinically relevant FVIII dose. Notably, a previous report showed that FVIII coadministered with OVA acted as an adjuvant inducing anti-OVA antibody response in HA mice, which otherwise did not respond to OVA alone.49 Future studies should provide more in-depth data on the effects of FVIII on DC phenotype in vivo and clarify whether these properties are a structural feature of FVIII, such as immunogenic posttranslational modifications, or a secondary effect of binding 1 of the several FVIII receptors or ligands. Moreover, our findings suggest that the pattern of antigen trafficking in the spleen represents another important factor in determining immunogenicity. RPMFs robustly took up OVA but not FVIII. Because RPMFs filter out and process large amounts of self-antigens, they can efficiently suppress helper T-cell responses to prevent autoimmunity.53 Perhaps failure to piggyback on that tolerogenic mechanism, in addition to induction of T-cell proliferation at low antigen doses as a result of efficient trafficking to DCs, helps seal the immunological fate of FVIII.
Acknowledgments
The authors thank Carol H. Miao for providing C57BL/6 hemophilia A mice.
This work was supported by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute grants P01 HL160472 and R01 HL133191 (R.W.H.) and U54 HL142012 (R.C. and R.W.H.), by Indiana Collaborative Initiative for Talent Enrichment (INCITE) funds (provided by Lilly Endowment), the Riley Children's Foundation (R.W.H.), a Bayer Hemophilia Award (R.K.), and the Indiana Diabetes Research Center grant P30 DK097512, which provides support for intravital microscopy studies at the Indiana University. The authors thank the members of the Indiana University Melvin and Bren Simon Cancer Center Flow Cytometry Resource Facility for their outstanding technical support. The Indiana University Melvin and Bren Simon Comprehensive Cancer Center Flow Cytometry Resource Facility is funded in part by NIH, National Cancer Institute grant P30 CA082709, and NIH, National Institute of Diabetes and Digestive and Kidney Diseases grant U54 DK106846 (Cooperative Center of Excellence in Hematology). The Flow Cytometry Resource Facility is further supported, in part, by NIH instrumentation grant 1S10D012270.
Authorship
Contribution: R.K., A.R.P., P.E.P., T.B., G.Q.P., A.S., J.B., M.C.A., M.M.K., M.M.M, S.M.Q., J.J.M., A.R.W., M.M.-M., and S.A. performed experiments; R.K., A.S., M.B., K.W.D., T.K., M.H.K., A.K.L., L.A.G., R.M.C., and R.W.H. designed the experiments; R.K. and R.W.H. analyzed and interpreted the data and wrote the manuscript; and R.W.H. supervised the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Roland W. Herzog, Herman B Wells Center for Pediatric Research, Indiana University, Indianapolis, IN; e-mail: rwherzog@iu.edu; and Radoslaw Kaczmarek, Herman B Wells Center for Pediatric Research, Indiana University, Indianapolis, IN; e-mail: rkaczmar@iu.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal