Key Points
LL37 primes platelet function and augments thrombus formation.
LL37 mainly acts through FPR2/ALX in platelets.

Abstract
Platelet-associated complications including thrombosis, thrombocytopenia, and hemorrhage are commonly observed during various inflammatory diseases such as sepsis, inflammatory bowel disease, and psoriasis. Despite the reported evidence on numerous mechanisms/molecules that may contribute to the dysfunction of platelets, the primary mechanisms that underpin platelet-associated complications during inflammatory diseases are not fully established. Here, we report the discovery of formyl peptide receptor 2, FPR2/ALX, in platelets and its primary role in the development of platelet-associated complications via ligation with its ligand, LL37. LL37 acts as a powerful endogenous antimicrobial peptide, but it also regulates innate immune responses. We demonstrate the impact of LL37 in the modulation of platelet reactivity, hemostasis, and thrombosis. LL37 activates a range of platelet functions, enhances thrombus formation, and shortens the tail bleeding time in mice. By utilizing a pharmacological inhibitor and Fpr2/3 (an ortholog of human FPR2/ALX)–deficient mice, the functional dependence of LL37 on FPR2/ALX was determined. Because the level of LL37 is increased in numerous inflammatory diseases, these results point toward a critical role for LL37 and FPR2/ALX in the development of platelet-related complications in such diseases. Hence, a better understanding of the clinical relevance of LL37 and FPR2/ALX in diverse pathophysiological settings will pave the way for the development of improved therapeutic strategies for a range of thromboinflammatory diseases.
Introduction
Platelets play pivotal roles in the regulation of hemostasis; however, their unwarranted activation under pathological conditions leads to the formation of blood clots (thrombosis) within the circulation, which is a major cause of premature death.1-3 Platelets also play significant roles in the regulation of innate immunity, inflammatory responses, and microbial infection.4,5 The activation of platelets during inflammatory diseases induces the formation of blood clots or disseminated intravascular coagulation in capillaries, resulting in the blockage of blood supply to tissues.6,7 Moreover, platelet activation results in the aggregation and sequestration of platelets, instigating thrombocytopenia.8,9 Several mechanisms that contribute to platelet dysfunction under inflammatory diseases have been reported10 ; however, the primary molecular mechanisms that underpin platelet activation are not fully understood.
LL37 is the only cathelicidin known to be expressed in human cells.11 It acts as a powerful antimicrobial peptide against bacteria,12 fungi,13 and viral particles14 and modulates innate and adaptive immune responses predominantly through formyl peptide receptor 2 (FPR2/ALX).15,16 Despite detailed research on the roles of LL37 in the modulation of inflammatory responses in various pathological settings,17,18 its effects in the regulation of thrombosis and other platelet-related complications remained unknown for a long time. Because the level of LL37 released during inflammation is significantly higher than normal,19,20 understanding its critical functions in the modulation of platelet reactivity will pave the way for determining fundamental mechanisms underlying platelet-related complications in various inflammatory diseases. Immediately prior to the submission of this manuscript, Pircher et al21 demonstrated the ability of LL37 to prime circulating platelets and induce thromboinflammation. Although they have established a significant functional impact of LL37 in platelets, its actions via FPR2/ALX were not determined. In this study, we report the effects of LL37 on a range of platelet functional assays and establish its roles in the modulation of platelet reactivity, thrombosis, and hemostasis. Moreover, by using pharmacological tools and Fpr2/3-deficient mice, we have established the functional dependence of LL37 on FPR2/ALX in platelets.
Methods
The University of Reading Research Ethics Committee approved all the experimental procedures using human blood from healthy volunteers. The mouse strains of Fpr1−/−22 and Fpr2/3−/−23 on a C57BL/6 background obtained from William Harvey Research Institute (London, United Kingdom) and control C57BL/6 mice from Envigo (United Kingdom) were used in this study. Detailed methods for the preparation of platelets, immunoblotting, enzyme-linked immunosorbent assay (ELISA), immunocytochemistry, in vitro thrombus formation, tail bleeding, platelet aggregation, dense granule secretion, platelet spreading, calcium mobilization, cytotoxicity and flow cytometry-based assays, mass spectrometry, molecular docking, and statistical analyses are provided in the supplemental Methods.
Results
Platelets store LL37 and release it upon activation
The expression of LL37 has been reported in several cell types including neutrophils where it is mainly stored in granules.24,25 By using immunofluorescence microscopy, we determined the presence of LL37 in human platelets (Figure 1A). Furthermore, resting and cross-linked collagen-related peptide (CRP-XL) (a glycoprotein VI [GPVI]-selective agonist) (1 μg/mL)–activated human platelets were centrifuged, and the supernatant and pellet were used in ELISA to determine the release of LL37 from platelets. In the resting state, the level of LL37 was significantly higher in the pellet (1559 ± 433 pM) compared with the supernatant (41 ± 12 pM) (Figure 1B). However, upon activation of platelets with CRP-XL, the presence of LL37 was significantly increased in the supernatant (1490 ± 581.7 pM) compared with the pellet (59 ± 7.6 pM). Similar results were obtained upon the activation of platelets with collagen (acts via GPVI and integrin α2β1) or TRAP-6 (acts via PAR1). To corroborate these results, the release of LL37 in human platelet-poor plasma (PPP) or platelet-rich plasma (PRP) was investigated by mass spectrometry. The level of LL37 was stable and significantly increased in PRP compared with PPP over 2 hours, indicating its release; by contrast, the level of LL37 was significantly reduced in PPP (Figure 1C). Together, these data confirm the presence of LL37 in platelets (between picomolar and nanomolar concentrations) and its release upon platelet activation.
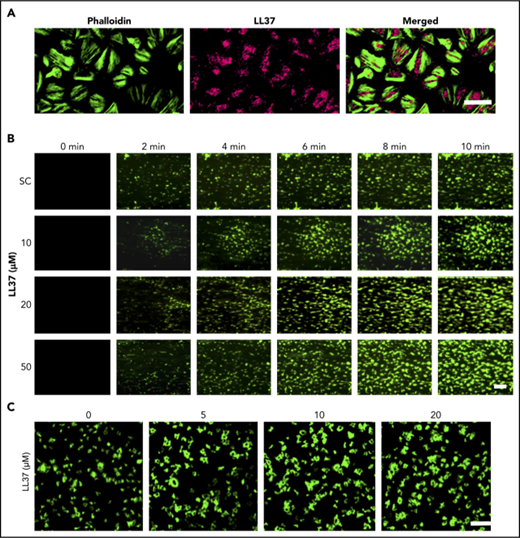
![Figure 1. Presence of LL37 in platelets and its impact on thrombus formation, hemostasis, and platelet activation. (A) Human platelets were treated with primary antibodies against LL37 and appropriate fluorescent-labeled secondary antibodies (purple) and phalloidin (green) and analyzed by confocal microscopy (magnification ×100; bar represents 10 µm). Images shown are representative of 3 independent experiments. (B) the level of LL37 in resting and activated (1 µg/mL CRP-XL, 1 µg/mL collagen or 10 µM thrombin receptor activator peptide 6 [TRAP-6]) platelet pellets and supernatants (SNs) was measured by ELISA using LL37-selective antibodies. Data represent mean ± standard error of the mean (SEM) (n = 5). (C) The stability/release of LL37 in human plasma was analyzed by mass spectrometry (liquid chromatography mass spectrometry [LC-MS]). Graph represents the intensities of LL37 (100 μg/mL) spiked in PRP and PPP at different time points over 120 minutes. Control represents the intensity of LL37 at 100 μg/mL (unspiked). Data represent mean ± SEM (n = 3). (D) The effects of LL37 in the modulation of thrombus formation. Human DiOC6-labeled whole blood was preincubated with a scrambled peptide (SC) or LL37 (10, 20, and 50 µM) for 10 minutes prior to perfusion over collagen-coated (400 µg/mL) Vena8 Biochips. Images (i) (at 10 minutes) shown are representative of 3 separate experiments (magnification ×10; bar represents 10 µm). Data (ii-iii) represent mean ± SEM (n = 3). (E) The impact of LL37 (20 µM) on the modulation of hemostasis. C57BL/6 mice (10-12 weeks old) were anesthetized 20 minutes before the infusion of a scrambled peptide or LL37 (20 µM) via femoral artery 5 minutes before the dissection of 1 mm of tail tip, and monitoring of time to cessation of bleeding. Data represent mean ± SEM (n = 6 per group). (F) The effects of LL37 on platelet activation were measured by optical aggregometry using human isolated platelets. Data represent mean ± SEM (n = 6). The statistical significance was established by 1-way analysis of variance (ANOVA) followed by Bonferroni’s correction in most of the experiments except the data shown in panels B and E, which were analyzed by 2-tailed unpaired Student t test and nonparametric Mann-Whitney U test, respectively (*P < .05; **P < .01).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/2/21/10.1182_bloodadvances.2018021758/4/m_advances021758f1.png?Expires=1769187136&Signature=ujgMqUXNm5vDZCKpFMZQxV7YUPyOyBvp4jkgegVeL9hMt5A7bnO3gpatjT6M5Z-zhW2FayELHfIe1zzRESysN8b0TsfqgsLpPi7QaMrAKczO~jimvxHEZHoX5bh1DHSPBIOL1QkM0zIkhthgRT5ucgkm6ZqT6KM8XYq2SG0We--B1JDe9MlBUak1fyJR8K9EnMmJsJyjneyykNwLZ9U3sFCjQv2v8gfVwNxQ90wJ7gIP69Stt88c9EG0kmR1De9oUldwBO9Onw-Vcvv8NvrEaPW6l7OwWhsvlZYJ9m6T9xEOtS8Q04-OvGTZBnIBomMXNtjaoY4KwvrrfhvwpHLiQA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Presence of LL37 in platelets and its impact on thrombus formation, hemostasis, and platelet activation. (A) Human platelets were treated with primary antibodies against LL37 and appropriate fluorescent-labeled secondary antibodies (purple) and phalloidin (green) and analyzed by confocal microscopy (magnification ×100; bar represents 10 µm). Images shown are representative of 3 independent experiments. (B) the level of LL37 in resting and activated (1 µg/mL CRP-XL, 1 µg/mL collagen or 10 µM thrombin receptor activator peptide 6 [TRAP-6]) platelet pellets and supernatants (SNs) was measured by ELISA using LL37-selective antibodies. Data represent mean ± standard error of the mean (SEM) (n = 5). (C) The stability/release of LL37 in human plasma was analyzed by mass spectrometry (liquid chromatography mass spectrometry [LC-MS]). Graph represents the intensities of LL37 (100 μg/mL) spiked in PRP and PPP at different time points over 120 minutes. Control represents the intensity of LL37 at 100 μg/mL (unspiked). Data represent mean ± SEM (n = 3). (D) The effects of LL37 in the modulation of thrombus formation. Human DiOC6-labeled whole blood was preincubated with a scrambled peptide (SC) or LL37 (10, 20, and 50 µM) for 10 minutes prior to perfusion over collagen-coated (400 µg/mL) Vena8 Biochips. Images (i) (at 10 minutes) shown are representative of 3 separate experiments (magnification ×10; bar represents 10 µm). Data (ii-iii) represent mean ± SEM (n = 3). (E) The impact of LL37 (20 µM) on the modulation of hemostasis. C57BL/6 mice (10-12 weeks old) were anesthetized 20 minutes before the infusion of a scrambled peptide or LL37 (20 µM) via femoral artery 5 minutes before the dissection of 1 mm of tail tip, and monitoring of time to cessation of bleeding. Data represent mean ± SEM (n = 6 per group). (F) The effects of LL37 on platelet activation were measured by optical aggregometry using human isolated platelets. Data represent mean ± SEM (n = 6). The statistical significance was established by 1-way analysis of variance (ANOVA) followed by Bonferroni’s correction in most of the experiments except the data shown in panels B and E, which were analyzed by 2-tailed unpaired Student t test and nonparametric Mann-Whitney U test, respectively (*P < .05; **P < .01).
Presence of LL37 in platelets and its impact on thrombus formation, hemostasis, and platelet activation. (A) Human platelets were treated with primary antibodies against LL37 and appropriate fluorescent-labeled secondary antibodies (purple) and phalloidin (green) and analyzed by confocal microscopy (magnification ×100; bar represents 10 µm). Images shown are representative of 3 independent experiments. (B) the level of LL37 in resting and activated (1 µg/mL CRP-XL, 1 µg/mL collagen or 10 µM thrombin receptor activator peptide 6 [TRAP-6]) platelet pellets and supernatants (SNs) was measured by ELISA using LL37-selective antibodies. Data represent mean ± standard error of the mean (SEM) (n = 5). (C) The stability/release of LL37 in human plasma was analyzed by mass spectrometry (liquid chromatography mass spectrometry [LC-MS]). Graph represents the intensities of LL37 (100 μg/mL) spiked in PRP and PPP at different time points over 120 minutes. Control represents the intensity of LL37 at 100 μg/mL (unspiked). Data represent mean ± SEM (n = 3). (D) The effects of LL37 in the modulation of thrombus formation. Human DiOC6-labeled whole blood was preincubated with a scrambled peptide (SC) or LL37 (10, 20, and 50 µM) for 10 minutes prior to perfusion over collagen-coated (400 µg/mL) Vena8 Biochips. Images (i) (at 10 minutes) shown are representative of 3 separate experiments (magnification ×10; bar represents 10 µm). Data (ii-iii) represent mean ± SEM (n = 3). (E) The impact of LL37 (20 µM) on the modulation of hemostasis. C57BL/6 mice (10-12 weeks old) were anesthetized 20 minutes before the infusion of a scrambled peptide or LL37 (20 µM) via femoral artery 5 minutes before the dissection of 1 mm of tail tip, and monitoring of time to cessation of bleeding. Data represent mean ± SEM (n = 6 per group). (F) The effects of LL37 on platelet activation were measured by optical aggregometry using human isolated platelets. Data represent mean ± SEM (n = 6). The statistical significance was established by 1-way analysis of variance (ANOVA) followed by Bonferroni’s correction in most of the experiments except the data shown in panels B and E, which were analyzed by 2-tailed unpaired Student t test and nonparametric Mann-Whitney U test, respectively (*P < .05; **P < .01).
LL37 augments thrombus formation and affects hemostasis
To determine whether LL37 has a direct influence on thrombotic complications during inflammatory diseases, its effects on thrombus formation under arterial flow conditions were investigated.26,27 LL37 (10, 20, and 50 µM) significantly increased the thrombus formation (Figure 1Di) and the mean fluorescence intensity in a concentration-dependent manner (Figure 1Dii). The highest concentration of LL37 (50 µM) increased the thrombus intensity by ∼70% compared with the vehicle-treated samples (Figure 1Diii). Moreover, LL37 affected hemostasis in mice as determined by a tail bleeding assay. A mean bleeding time of 370.8 ± 46.6 seconds was observed in the vehicle-treated group; however, the infusion of LL37 significantly shortened the bleeding time to a mean of 225.2 ± 18.8 seconds (Figure 1E).
LL37 induces platelet activation
To further determine the impact of LL37 in distinctive platelet functions, additional assays were performed. Human isolated platelets were treated with a vehicle or LL37 (5, 10, and 20 µM), and the level of aggregation was monitored by optical aggregometry. LL37 directly induced platelet aggregation in a concentration-dependent manner, notably, 20 µM LL37 induced 100% aggregation (Figure 1F). In line with a recent study,21 in our experiments, LL37 failed to induce aggregation in human PRP (supplemental Figure 1). Similarly, to determine whether LL37 influences inside-out signaling to integrin αIIbβ3 and α-granule secretion, the level of fibrinogen binding and P-selectin exposure was measured respectively using human isolated platelets and PRP by flow cytometry. LL37 increased the level of fibrinogen binding in human isolated platelets (Figure 2Ai) and PRP (Figure 2Aii) in a concentration-dependent manner. Similarly, the level of P-selectin exposure was increased by LL37 in isolated platelets (Figure 2Bi) and PRP (Figure 2Bii). Moreover, the ability of LL37 to induce platelet spreading on immobilized fibrinogen (Figure 2C) was analyzed as a marker for integrin αIIbβ3-mediated outside-in signaling. LL37 (5, 10, and 20 µM) significantly increased the number of adhered (Figure 2Ci) and spread (Figure 2Cii) platelets, and the relative surface area (Figure 2Ciii) compared with their controls. Furthermore, to assess the effects of LL37 in calcium mobilization, intracellular calcium levels were measured in human isolated platelets. LL37 induced calcium mobilization with a maximum level achieved with 50 µM. The level of calcium release obtained with 50 µM LL37 is similar to that obtained with CRP-XL (1 µg/mL), although the initial kinetics of calcium release appeared to be faster for LL37 (Figure 2D). These data confirm that LL37 triggers distinctive platelet functions.

The impact of LL37 on platelet activation, spreading, and calcium mobilization. (A) The level of fibrinogen binding was analyzed using fluorescein isothiocyanate–conjugated fibrinogen antibodies by flow cytometry in human isolated platelets (i) or PRP (ii). (B) Similarly, the level of P-selectin exposure was measured in human isolated platelets (i) or PRP (ii) using PECy5-labeled P-selectin antibodies. Data represent mean ± SEM (n = 3). (C) Platelet adhesion and spreading on immobilized fibrinogen was analyzed using platelets treated with LL37 (5, 10, and 20 µM) by confocal microscopy (magnification ×60; bar represents 10 µm). The number of adhered (i) and spread (ii) platelets, and the relative surface area of spread platelets (iii) was determined via analyzing the images using ImageJ. Ten random fields of view were recorded and analyzed for each sample. Data represent mean ± SEM (n = 3). (D) Ca2+ mobilization was measured using Fluo-4 am dye-loaded human isolated platelets upon stimulation with LL37 by spectrofluorimetry. Data represent mean of maximum level of Ca2+ ± SEM (n = 4). (E) The cytotoxic effects of LL37 were measured in human isolated platelets using a lactate dehydrogenase cytotoxicity assay kit. Data represent mean ± SEM (n = 4). The statistical significance was established by 1-way ANOVA followed by Bonferroni’s correction (*P < .05; **P < .01; ***P < .001).
The impact of LL37 on platelet activation, spreading, and calcium mobilization. (A) The level of fibrinogen binding was analyzed using fluorescein isothiocyanate–conjugated fibrinogen antibodies by flow cytometry in human isolated platelets (i) or PRP (ii). (B) Similarly, the level of P-selectin exposure was measured in human isolated platelets (i) or PRP (ii) using PECy5-labeled P-selectin antibodies. Data represent mean ± SEM (n = 3). (C) Platelet adhesion and spreading on immobilized fibrinogen was analyzed using platelets treated with LL37 (5, 10, and 20 µM) by confocal microscopy (magnification ×60; bar represents 10 µm). The number of adhered (i) and spread (ii) platelets, and the relative surface area of spread platelets (iii) was determined via analyzing the images using ImageJ. Ten random fields of view were recorded and analyzed for each sample. Data represent mean ± SEM (n = 3). (D) Ca2+ mobilization was measured using Fluo-4 am dye-loaded human isolated platelets upon stimulation with LL37 by spectrofluorimetry. Data represent mean of maximum level of Ca2+ ± SEM (n = 4). (E) The cytotoxic effects of LL37 were measured in human isolated platelets using a lactate dehydrogenase cytotoxicity assay kit. Data represent mean ± SEM (n = 4). The statistical significance was established by 1-way ANOVA followed by Bonferroni’s correction (*P < .05; **P < .01; ***P < .001).
LL37 does not exhibit cytotoxic effects in platelets
A previous study28 reported the inhibitory effects of LL37 in platelets at exceptionally high concentrations (0.1-1.2 mM). It has been shown previously that LL37 exhibits cytotoxic effects in neutrophils and monocytes at concentrations higher than 50 µM.29 To determine whether the concentrations of LL37 used in this study (≤50 µM) exhibit any cytotoxic effects in platelets, lactate dehydrogenase cytotoxicity assay was performed. The concentrations of LL37 used here (1-50 µM) failed to exert any cytotoxic effects in human isolated platelets, although 100 µM LL37 displayed significant toxicity (Figure 2E). These results confirm that LL37 concentrations up to 50 µM do not display any toxic effects in platelets, although higher concentrations may exhibit cytotoxic effects.
LL37 activates platelets selectively through FPR2/ALX
Numerous studies indicate that LL37 acts primarily through FPR2/ALX to exert its effects in immune cells.16,30,31 The expression of FPR2/ALX in megakaryocytes and human and mouse platelets at the transcript level has been reported previously.32,33 Here, the presence of FPR2/ALX in human platelets remained unchanged in resting and CRP-XL (1 μg/mL)–activated platelets as confirmed by immunoblots (Figure 3Ai), although the activation increased the surface level as determined by flow cytometry (Figure 3Aii). Moreover, the presence of Fpr2/3 (an ortholog to human FPR2/ALX) in control, and its absence in Fpr2/3−/− mouse platelets, was confirmed (Figure 3Aiii).

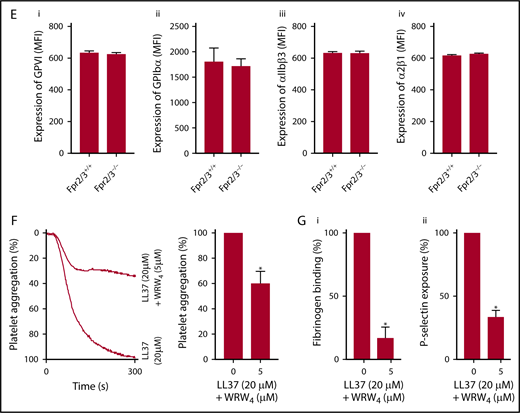
Expression of FPR2/ALX in platelets and its influence on LL37-mediated platelet activation. The presence of FPR2/ALX was confirmed in human (Ai) and mouse (Aiii) platelet lysates by immunoblot analysis using selective antibodies. The blots are representative of 3 separate experiments. The expression of FPR2/ALX on the surface of resting or activated (1 µg/mL CRP-XL) human platelets was analyzed using FPR2/ALX-selective and fluorescent-labeled secondary antibodies by flow cytometry (Aii). Data represent mean ± SEM (n = 4). The binding of LL37 to platelets was analyzed by flow cytometry. Human isolated platelets were incubated with 20 μM 5-FAM-LC-conjugated LL37 (FL-LL37) or scrambled LL37 (FL-scLL37), and the level of binding was analyzed by flow cytometry (Bi). Data represent mean ± SEM (n = 4). Similarly, platelets obtained from control, Fpr2/3−/−, and Fpr1−/− mice were analyzed with 20 μM 5-FAM-LC-conjugated LL37 or scrambled 5-FAM-LC-LL37 (Bii). Data represent mean ± SEM (n = 7). Similarly, platelets obtained from control or Fpr2/3−/− mice were analyzed with 20 μM 5-FAM-conjugated mCRAMP or scrambled 5-FAM-mCRAMP (Biii). Data represent mean ± SEM (n = 4). The interactions between LL37 and FPR2/ALX were analyzed through structural modeling and molecular docking analysis (Biv). (C) The level of fibrinogen binding upon stimulation with LL37 in isolated platelets (i) or whole blood (ii) obtained from Fpr2/3−/− or control mice was analyzed by flow cytometry. Data represent mean ± SEM (n = 10 for panel Ci; n = 8 for panel Cii). (D) Similarly, the level of P-selectin exposure was analyzed using isolated platelets (i) or whole blood (ii) from these mice. Data represent mean ± SEM (n = 10 for panel Di; n = 13 for panel Dii). (E) The expression levels of major platelet receptors such as GPVI (i), GPIbα (ii), αIIbβ3 (iii), and α2β1 (iv) in platelets obtained from Fpr2/3−/− and control mice were analyzed by flow cytometry using selective fluorescent-labeled antibodies. Data represent mean ± SEM (n = 8 per group). (F) The effect of a selective inhibitor for FPR2/ALX, WRW4 (5 µM), on LL37-induced platelet activation was measured by optical aggregometry. Data represent mean ± SEM (n = 3). (G) Mouse isolated platelets were stimulated with LL37 (20 μM) in the presence or absence WRW4 (5 μM), and the level of fibrinogen binding (i) and P-selectin exposure (ii) were analyzed by flow cytometry. Data represent mean ± SEM (n = 4). The statistical significance was calculated using 1-way ANOVA followed by Bonferroni’s correction in most of the experiments except for the data shown in panels Aii, B, and E-G, where a 2-tailed unpaired Student t test was used (*P < .05; **P < .001; ***P < .0001).
Expression of FPR2/ALX in platelets and its influence on LL37-mediated platelet activation. The presence of FPR2/ALX was confirmed in human (Ai) and mouse (Aiii) platelet lysates by immunoblot analysis using selective antibodies. The blots are representative of 3 separate experiments. The expression of FPR2/ALX on the surface of resting or activated (1 µg/mL CRP-XL) human platelets was analyzed using FPR2/ALX-selective and fluorescent-labeled secondary antibodies by flow cytometry (Aii). Data represent mean ± SEM (n = 4). The binding of LL37 to platelets was analyzed by flow cytometry. Human isolated platelets were incubated with 20 μM 5-FAM-LC-conjugated LL37 (FL-LL37) or scrambled LL37 (FL-scLL37), and the level of binding was analyzed by flow cytometry (Bi). Data represent mean ± SEM (n = 4). Similarly, platelets obtained from control, Fpr2/3−/−, and Fpr1−/− mice were analyzed with 20 μM 5-FAM-LC-conjugated LL37 or scrambled 5-FAM-LC-LL37 (Bii). Data represent mean ± SEM (n = 7). Similarly, platelets obtained from control or Fpr2/3−/− mice were analyzed with 20 μM 5-FAM-conjugated mCRAMP or scrambled 5-FAM-mCRAMP (Biii). Data represent mean ± SEM (n = 4). The interactions between LL37 and FPR2/ALX were analyzed through structural modeling and molecular docking analysis (Biv). (C) The level of fibrinogen binding upon stimulation with LL37 in isolated platelets (i) or whole blood (ii) obtained from Fpr2/3−/− or control mice was analyzed by flow cytometry. Data represent mean ± SEM (n = 10 for panel Ci; n = 8 for panel Cii). (D) Similarly, the level of P-selectin exposure was analyzed using isolated platelets (i) or whole blood (ii) from these mice. Data represent mean ± SEM (n = 10 for panel Di; n = 13 for panel Dii). (E) The expression levels of major platelet receptors such as GPVI (i), GPIbα (ii), αIIbβ3 (iii), and α2β1 (iv) in platelets obtained from Fpr2/3−/− and control mice were analyzed by flow cytometry using selective fluorescent-labeled antibodies. Data represent mean ± SEM (n = 8 per group). (F) The effect of a selective inhibitor for FPR2/ALX, WRW4 (5 µM), on LL37-induced platelet activation was measured by optical aggregometry. Data represent mean ± SEM (n = 3). (G) Mouse isolated platelets were stimulated with LL37 (20 μM) in the presence or absence WRW4 (5 μM), and the level of fibrinogen binding (i) and P-selectin exposure (ii) were analyzed by flow cytometry. Data represent mean ± SEM (n = 4). The statistical significance was calculated using 1-way ANOVA followed by Bonferroni’s correction in most of the experiments except for the data shown in panels Aii, B, and E-G, where a 2-tailed unpaired Student t test was used (*P < .05; **P < .001; ***P < .0001).
To determine the functional dependence of LL37 on FPR2/ALX, the binding of LL37 to the platelet surface was confirmed using a fluorescently labeled LL37 (5-FAM-LC-conjugated LL37) by flow cytometry. 5-FAM-LL37 (20 μM) displayed marked binding to the surface of human platelets compared with a fluorescently labeled scrambled LL37 (5-FAM-conjugated sc-LL37) (Figure 3Bi). Similarly, LL37 (20 μM) binding was analyzed using platelets obtained from control, Fpr1-, or Fpr2/3-deficient mice. The control and Fpr1−/− mouse platelets exhibited significant binding to 5-FAM-LL37 compared with Fpr2/3−/− (Figure 3Bii). In addition to LL37, mCRAMP (mouse ortholog of LL37) was also investigated for the binding to mouse platelets, and indeed, the level of mCRAMP binding was reduced in Fpr2/3−/− mouse platelets compared with the controls (Figure 3Biii). The interactions between LL37 and FPR2/ALX were examined by molecular docking analysis, which predicted that LL37 peptide forms prominent hydrogen bonds with key residues such as Gln-89, Ser-182, Asn-285, and Gly-275 of FPR2/ALX that are identified as crucial for receptor activation (Figure 3Biv; Table 1).
The functional dependence of LL37 on FPR2/ALX was analyzed by using a range of platelet functional assays. The activatory effects of LL37 (5-50 µM) were substantially reduced in Fpr2/3−/− mouse platelets both in isolation (Figure 3Ci,Di) or whole blood (Figure 3Cii,Dii) compared with the controls as analyzed by fibrinogen binding and P-selectin exposure. Notably, the characterization of platelets obtained from Fpr2/3−/− mice failed to display any defects in size and number of platelets or the levels of major platelet receptors such as GPVI (Figure 3Ei), glycoprotein Ibα (GPIbα) (Figure 3Eii), αIIbβ3 (Figure 3Eiii), and α2β1 (Figure 3Eiv) compared with the control mouse platelets. To corroborate these results, platelet functional assays were performed in the presence of a selective FPR2/ALX antagonist, WRW4 (WRWWWW), in human and mouse platelets. The addition of WRW4 (5 µM) in human isolated platelets before activation with 20 µM LL37 inhibited platelet aggregation by ∼40% (Figure 3F). Similarly, the effects of LL37 (20 µM) on fibrinogen binding (Figure 3Gi) and P-selectin exposure (Figure 3Gii) were significantly reduced in WRW4 (5 µM)–treated mouse platelets. These data demonstrate the involvement of FPR2/ALX in the regulation of LL37-mediated effects in platelets.
FPR2/ALX regulates normal platelet activation
To validate the importance of FPR2/ALX in the regulation of normal platelet activation, further experiments were performed using human isolated platelets in the presence or absence of WRW4. CRP-XL (0.25 µg/mL)–induced platelet aggregation was significantly reduced in the presence of WRW4 (2.5-20 μM). The inhibition of FPR2/ALX with WRW4 (20 µM) reduced the platelet aggregation by ∼89% (Figure 4A). Similar results were obtained with adenosine 5′-diphosphate (ADP)–induced platelet aggregation, wherein WRW4 (20 µM) inhibited 75% of aggregation (Figure 4B). Moreover, dense granule secretion (evidenced by adenosine triphosphate release) was significantly reduced in the presence of WRW4 (Figure 4C). The platelet activation was also assessed using whole blood obtained from control and Fpr2/3−/− mice upon stimulation with conventional platelet agonists such as CRP-XL, ADP, AY-NH2 (activates protease activated receptor, PAR4), and U46619, an analog of thromboxane A2 by measuring the levels of fibrinogen binding and P-selectin exposure. Similar to human platelets, the activation of platelets obtained from Fpr2/3−/− mice upon stimulation with CRP-XL (Figure 4D), ADP (Figure 4E), AY-NH2 (Figure 4F), and U46619 (Figure 4G) was significantly reduced compared with the controls. Additionally, preincubation of human platelets with WRW4 (1.25-20 μM) significantly decreased the number of adhered (Figure 5Ai) and spread (Figure 5Aii) platelets, and the relative surface area (Figure 5Aiii). The impact of Fpr2/3 on the modulation of hemostasis in mice was determined by tail bleeding assay. A mean bleeding time of 428.5 ± 64.8 seconds was observed in the control group; however, Fpr2/3-deficient mice significantly increased the bleeding time to a mean of 1128 ± 71.9 seconds (Figure 5B). Together, these data emphasize the impact of FPR2/ALX on the regulation of normal platelet function through a positive feedback mechanism (may be through LL37), and thus the inhibition or deletion of this receptor results in diminished platelet function in general.

Positive feedback regulation of FPR2/ALX in platelets. The effects of different concentrations of WRW4 on CRP-XL– (0.25 µg/mL) (A) or ADP-induced (2 µM) (B) human platelet aggregation was analyzed by optical aggregometry. (C) The level of adenosine triphosphate (ATP) secretion in human platelets (PRP) treated with WRW4 prior to activation with CRP-XL (0.25 µg/mL) was measured by lumi-aggregometry. The levels of fibrinogen binding (i) and P-selectin exposure (ii) were analyzed in platelets obtained from control or Fpr2/3−/− mice upon stimulation with various concentrations of CRP-XL (D), ADP (E), AY-NH2 (F), or U46691 (G) by flow cytometry. Data represent mean ± SEM (n = 3). P values shown are as calculated by 2-way ANOVA followed by Bonferroni's correction in most of the experiments except for the data shown in panels A-C, which were analyzed by 1-way ANOVA followed by Bonferroni's correction (*P < .05; **P < .01).
Positive feedback regulation of FPR2/ALX in platelets. The effects of different concentrations of WRW4 on CRP-XL– (0.25 µg/mL) (A) or ADP-induced (2 µM) (B) human platelet aggregation was analyzed by optical aggregometry. (C) The level of adenosine triphosphate (ATP) secretion in human platelets (PRP) treated with WRW4 prior to activation with CRP-XL (0.25 µg/mL) was measured by lumi-aggregometry. The levels of fibrinogen binding (i) and P-selectin exposure (ii) were analyzed in platelets obtained from control or Fpr2/3−/− mice upon stimulation with various concentrations of CRP-XL (D), ADP (E), AY-NH2 (F), or U46691 (G) by flow cytometry. Data represent mean ± SEM (n = 3). P values shown are as calculated by 2-way ANOVA followed by Bonferroni's correction in most of the experiments except for the data shown in panels A-C, which were analyzed by 1-way ANOVA followed by Bonferroni's correction (*P < .05; **P < .01).

Impact of FPR2/ALX in platelet spreading, hemostasis, and cyclic adenosine monophosphate (cAMP)–mediated signaling. (A) Platelet adhesion and spreading on fibrinogen-coated glass surface was analyzed in the absence and presence of WRW4 (1.25, 2.5, 5, and 20 µM) by confocal microscopy (magnification ×60; bar indicates 10 µm). The number of adhered (Ai) and spread (Aii) platelets and the relative surface area of spread platelets (Aiii) were determined by analyzing the images using ImageJ. Ten random fields of view were recorded for each sample. Data represent mean ± SEM (n = 3). (B) The impact of FPR2/ALX in the modulation of hemostasis was analyzed using tail bleeding assay in control or Fpr2/3−/− mice. Data represent mean ± SEM (n = 8 in each group). (C) The level of cAMP in human isolated platelets in the presence or absence of WRW4 (i) and control and Fpr2/3−/− mouse platelets (ii) was analyzed using a cAMP assay kit. Data represent mean ± SEM (n = 4). (D) The phosphorylation of VASP at Ser-157 was analyzed in the presence of WRW4 (i) and in platelets obtained from Fpr2/3−/− mice (ii) by immunoblot analysis using selective antibodies. The blots are representative of 3 separate experiments. P values shown are as calculated by 1-way ANOVA followed by Bonferroni's correction in most of the experiments except for the data shown in panels B-C, where a nonparametric Mann-Whitney U test and a 2-tailed unpaired Student t test were used, respectively (*P < .05; **P < .01; ***P < .001).
Impact of FPR2/ALX in platelet spreading, hemostasis, and cyclic adenosine monophosphate (cAMP)–mediated signaling. (A) Platelet adhesion and spreading on fibrinogen-coated glass surface was analyzed in the absence and presence of WRW4 (1.25, 2.5, 5, and 20 µM) by confocal microscopy (magnification ×60; bar indicates 10 µm). The number of adhered (Ai) and spread (Aii) platelets and the relative surface area of spread platelets (Aiii) were determined by analyzing the images using ImageJ. Ten random fields of view were recorded for each sample. Data represent mean ± SEM (n = 3). (B) The impact of FPR2/ALX in the modulation of hemostasis was analyzed using tail bleeding assay in control or Fpr2/3−/− mice. Data represent mean ± SEM (n = 8 in each group). (C) The level of cAMP in human isolated platelets in the presence or absence of WRW4 (i) and control and Fpr2/3−/− mouse platelets (ii) was analyzed using a cAMP assay kit. Data represent mean ± SEM (n = 4). (D) The phosphorylation of VASP at Ser-157 was analyzed in the presence of WRW4 (i) and in platelets obtained from Fpr2/3−/− mice (ii) by immunoblot analysis using selective antibodies. The blots are representative of 3 separate experiments. P values shown are as calculated by 1-way ANOVA followed by Bonferroni's correction in most of the experiments except for the data shown in panels B-C, where a nonparametric Mann-Whitney U test and a 2-tailed unpaired Student t test were used, respectively (*P < .05; **P < .01; ***P < .001).
FPR2/ALX exerts its effects through cAMP-dependent signaling
FPRs are Gi-coupled receptors,34 which are known to inhibit adenylate cyclase, reducing the level of cAMP, a known inhibitor of platelet function. Therefore, the deletion of Gi-coupled receptor genes in mice increases the basal cAMP levels in target cells.35,36 To investigate whether the inhibition of FPR2/ALX in human or deletion of Fpr2/3 in mouse platelets is influenced by the cAMP-dependent signaling, the level of cAMP was quantified in platelets. The inhibition of FPR2/ALX with WRW4 (20 μM) significantly elevated the cAMP levels compared with the controls in human platelets (Figure 5Ci). Similarly, resting Fpr2/3−/− mouse platelets exhibited elevated basal levels of cAMP compared with the controls (Figure 5Cii). In order to corroborate this data, we investigated the phosphorylation of the vasodilator-stimulated phosphoprotein (VASP), a substrate for protein kinase A that is involved in the regulation cAMP-mediated signaling. The treatment of platelets with an FPR2/ALX-selective inhibitor, WRW4, increased the phosphorylation of Ser157-VASP (Figure 5Di). Similarly, Fpr2/3−/− mouse platelets demonstrated increased phosphorylation of Ser157-VASP compared with control mouse platelets (Figure 5Dii).
Discussion
LL37 is a powerful antimicrobial peptide that plays substantial roles in the initiation of chemotaxis and subsequent inflammatory responses in immune cells including monocytes,16 mast cells,37 eosinophils, and neutrophils.38 Moreover, the involvement of LL37 in the development of pathological conditions such as sepsis, inflammatory bowel disease, psoriasis, and cystic fibrosis has been previously reported, and hence, its therapeutic potential has been analyzed in detail.39 LL37 has also been found to play a role in the pathogenesis of atherosclerosis,40,41 wherein it leads to the development of lesions and recruitment of inflammatory cells at the site of injury.18 However, the role of LL37 in the modulation of thrombosis and hemostasis has not been investigated until recently. Immediately prior to the submission of this manuscript, a recent study demonstrated the functional impact of LL37 in priming platelets and inducing thromboinflammatory conditions, although the molecular mechanisms that regulate such effects were not fully established.21 However, in this study, we have investigated the impact of LL37 in the modulation of platelet reactivity, thrombosis, and hemostasis under physiological conditions and uncovered the functional dependence of LL37 on FPR2/ALX in platelets.
The expression of LL37 has been reported in numerous cell types including epithelial cells and immune cells such as neutrophils and monocytes.42 Because platelets are also derived from the myeloid lineage,43 we hypothesized that platelets may possess LL37, and indeed the presence of LL37 in platelets and its release to the external milieu upon activation were confirmed in this study. Similar to neutrophils, platelets may also contain LL37 in their granules and release it upon stimulation to increase its concentration at the local environment and enhance the secondary activation of platelets toward augmentation of thrombosis. A recent study21 also reported the elevated level of LL37 in the microenvironment of arterial thrombi in human and mice. Platelets are known to contain several antimicrobial peptides and release them upon activation during microbial infection.44 Similarly, platelets may contribute to the release of LL37 upon activation to support the microbial clearance, activation of inflammatory responses, and modulation of thrombosis and hemostasis during pathological settings. Although the activation of platelets during inflammatory diseases is inevitable because of the presence of several molecules that activate platelets,45 here we demonstrate LL37 as a major contributor to platelet activation and thrombus formation.
Thrombosis and subsequent bleeding are associated with various inflammatory diseases.46 Similarly, disseminated intravascular coagulation, thrombosis in the microvasculature, and sequestration of platelets are some of the common clinical manifestations in sepsis.47 The level of LL37 is significantly increased in psoriasis48 and sepsis49 patients compared with healthy individuals. In line with thrombosis in vasculature, LL37 augmented in vitro thrombus formation and shortened the bleeding time in mice. These data demonstrate a fundamental function for LL37 in the modulation of thrombosis and hemostasis. Similarly, LL37 induced platelet aggregation, fibrinogen binding, granule secretion, adhesion, spreading, and intracellular calcium mobilization in platelets. Pircher et al21 also demonstrated the functional impact of LL37 on arterial thrombosis and platelet granule secretion. In line with this previous study, we also did not observe the ability of LL37 to induce platelet aggregation in PRP, although it displayed aggregation in human isolated platelets. Similarly, the previous study has failed to detect a response for LL37 on integrin αIIbβ3 activation and platelet spreading on a fibrinogen-coated surface. In our study, however, we demonstrate the ability of LL37 to increase fibrinogen binding and platelet spreading. Surprisingly, the previous study failed to detect integrin αIIbβ3 activation. However, we have directly measured the level of fibrinogen binding in isolated platelets, PRP, and whole blood with a range of concentrations of LL37 (ie, up 50 μM), whereas they have measured PAC-1 binding in isolated platelets using LL37 concentrations of only up to 5 μM. It is unclear why these discrepancies occur between the previously reported data and our results, although it may be attributed in part to the differences in the methodologies used.
The concentrations used in this study (up to 50 µM) revealed the activatory effects of LL37 in platelets. A recent study reported the inhibitory effects of LL37 in platelets at concentrations between 0.1 and 1.2 mM.28 These concentrations are not only substantially greater than those achievable in pathological conditions, but also exert cytotoxic effects in several cell types29 including platelets (Figure 2E) at 100 µM. However, in severe psoriasis, a median concentration of 304 µM LL37 in psoriatic lesions has been reported,48 which can exert cytotoxicity towards platelets at the local sites and reduce the number of functional platelets. During pulmonary infection, a concentration of 5 µM LL37 has been detected.50 Notably, the normal plasma concentration of LL37 in healthy individuals is suggested to be ∼1.2 µM,51 which did not exert any effects on platelets. The LL37 concentrations used in this study are similar to those achievable during pathological conditions, such as sepsis,49 and early stages of psoriasis. Hence, the LL37 inhibitory effects previously reported in platelets may well be because of cytotoxicity, although additional causes cannot be excluded. This perspective was also reflected in the recent study by Pircher et al.21 Together with the previous reports, our data demonstrate that LL37 induces platelet activation at the early stages of inflammatory diseases resulting in the initiation of thrombosis and modulation of hemostasis. However, at concentrations of 100 µM and above, LL37 may exert cytotoxicity, reducing the number and function of circulating platelets, which can ultimately lead to bleeding complications. Together with the modulation of thrombosis and hemostasis, LL37 may also induce other platelet-related complications (eg, thrombocytopenia and inflammation) during various inflammatory diseases where its level is elevated. Notably, the previous study21 has demonstrated the impact of LL37 in the augmentation of platelet-neutrophil interactions, cytokine release, release of extracellular nucleosomes, and reactive oxygen species.
LL37 has been reported to act mainly through FPR2/ALX in other cell types,16,24 although additional receptors such as Toll-like receptors, receptor tyrosine kinases, ligand-gated ion channel, CCR3, P2Y11, and P2X7 were shown to bind this peptide.52 To investigate the underlying molecular mechanisms through which LL37 modulates platelet function, the effects of LL37 in human platelets treated with WRW4 and platelets obtained from Fpr2/3−/− mice were analyzed. LL37-mediated activation was largely reduced by WRW4 and in platelets obtained from Fpr2/3−/− mice, confirming the functional dependence of LL37 primarily on FPR2/ALX. LL37 binding assays confirmed the substantial reduction in the binding of LL37 to Fpr2/3−/− platelet surface compared with Fpr1−/− and control mouse platelets. Additionally, we were able to demonstrate that FPR2/ALX inhibition in human platelets or deletion of Fpr2/3 gene in mice lead to the elevation of cAMP levels, which is a major inhibitory molecule for platelet activation. This indicates the involvement of cAMP-dependent signaling pathways in the regulation of FPR2/ALX in platelets. Nevertheless, the activation of platelets by LL37 through receptors other than FPRs cannot be excluded, and further investigations will be needed to explore the contributions of such receptors in the modulation of platelet function upon ligation with LL37. Interestingly, the recent study21 has reported that LL37 is partially acting through the GPVI signaling pathway in platelets, but it does not have any impact on G protein-coupled receptor–mediated signaling specifically via FPR1 and FPR2/ALX using a single concentration of pharmacological inhibitors (Boc-MLF and WRW4) for these receptors. However, in the present study, we report the presence of FPR2/ALX (a G protein-coupled receptor) and its role in the regulation of LL37-mediated effects in platelets using a range of concentrations of these pharmacological inhibitors and platelets obtained from Fpr2/3−/− mice in multiple experiments.
In conclusion, we demonstrate that LL37 is stored in platelets and secreted upon activation. Similar to a recent publication,21 we also report that LL37 promotes thrombus formation. LL37 induced a range of platelet function such as platelet aggregation, inside-out signaling to integrin αIIbβ3 and outside-in signaling, granule secretion, calcium mobilization, and shortened tail bleeding time in mice. These effects were diminished in the presence of an FPR2/ALX pharmacological inhibitor and in platelets obtained from Fpr2/3−/− mice confirming the functional dependence of LL37 primarily via this receptor in platelets. Additionally, we demonstrate an instrumental role for FPR2/ALX in the positive feedback regulation of platelet function, in which the deficiency or blockade of this receptor impaired hemostasis and the normal activation of platelets. The significant roles of LL37 and FPR2/ALX in the modulation of thrombosis and hemostasis renders them potential candidates for the exacerbation of platelet-related complications and immune responses in numerous inflammatory diseases where platelets play critical roles. Notably, the presence of FPRs in platelets opens up new avenues to investigate the involvement of a multiplicity of FPR ligands in the modulation of thrombosis, hemostasis, and other platelet-related complications during inflammatory responses. Based on the data presented in this study, both LL37 and FPRs can act as potential therapeutic targets for cardiovascular and a range of inflammatory diseases.
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by grants from the Wellcome Trust (204389/Z/16/Z), the British Heart Foundation (PG/16/64/32311), the Royal Society, the British Pharmacological Society, the Physiological Society, and the Saudi Arabian Ministry of Higher Education.
Authorship
Contribution: M.F.S. and S.V. designed the study, performed experiments, analyzed data, and wrote the manuscript; D.R., X.K., L.A.M., H.F.W., T.M.V., D.A.A., R.V., and K.W. performed experiments and analyzed data; and J.M.G., S.D.B., and M.P. provided expertise and reagents for specific experiments and support during the preparation of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sakthivel Vaiyapuri, School of Pharmacy, University of Reading, Reading RG6 6UB, United Kingdom; e-mail: s.vaiyapuri@reading.ac.uk.