

Key Points
Platelet CD36 signaling promotes caspase activity through redox sensor MAPK ERK5.
ERK5 and caspases link platelet CD36 to fibrin accumulation by exposing surface procoagulant PSer in dyslipidemic conditions.

Abstract
Dyslipidemia is a risk factor for clinically significant thrombotic events. In this condition, scavenger receptor CD36 potentiates platelet reactivity through recognition of circulating oxidized lipids. CD36 promotes thrombosis by activating redox-sensitive signaling molecules, such as the MAPK extracellular signal-regulated kinase 5 (ERK5). However, the events downstream of platelet ERK5 are not clear. In this study, we report that oxidized low-density lipoprotein (oxLDL) promotes exposure of procoagulant phosphatidylserine (PSer) on platelet surfaces. Studies using pharmacologic inhibitors indicate that oxLDL-CD36 interaction–induced PSer exposure requires apoptotic caspases in addition to the downstream CD36-signaling molecules Src kinases, hydrogen peroxide, and ERK5. Caspases promote PSer exposure and, subsequently, recruitment of the prothrombinase complex, resulting in the generation of fibrin from the activation of thrombin. Caspase activity was observed when platelets were stimulated with oxLDL. This was prevented by inhibiting CD36 and ERK5. Furthermore, oxLDL potentiates convulxin/glycoprotein VI–mediated fibrin formation by platelets, which was prevented when CD36, ERK5, and caspases were inhibited. Using 2 in vivo arterial thrombosis models in apoE-null hyperlipidemic mice demonstrated enhanced arterial fibrin accumulation upon vessel injury. Importantly, absence of ERK5 in platelets or mice lacking CD36 displayed decreased fibrin accumulation in high-fat diet–fed conditions comparable to that seen in chow diet–fed animals. These findings suggest that platelet signaling through CD36 and ERK5 induces a procoagulant phenotype in the hyperlipidemic environment by enhancing caspase-mediated PSer exposure.
Introduction
Dyslipidemia is a risk factor for clinically significant arterial thrombosis, a major cause of heart attack and stroke. In this setting, thrombosis is initiated by activation of blood platelets and the coagulation cascade after exposure to plaque contents and subendothelial tissue factor.1 In dyslipidemia, subthreshold levels of platelet activation can potentiate these early thrombotic events thus increasing the risk of life-threatening occlusive thrombosis. Micromolar levels of oxidized lipids, circulating within low-density lipoprotein (LDL) particles (oxidized LDL [oxLDL]) and generated from the oxidative processes of plaque formation,2 lower the threshold for platelet activation through specific pattern recognition receptors, including CD36.3
CD36 is highly expressed on the surface of platelets. Expression levels vary substantially in the human populations and have been linked to specific polymorphisms associated with risk of myocardial infarction.4 In dyslipidemia, CD36 recognizes oxLDL and potentiates platelet activation.2,5 This is through activation of multiple signaling pathways, including Src family kinases Fyn and Lyn6 and nonreceptor tyrosine kinase Syk7,8 ; Vav family guanine nucleotide exchange factors9 ; the phospholipase Cγ2–protein kinase C (PKC)–nicotinamide adenine dinucleotide phosphate (NADPH) oxidase signaling axis that generates reactive oxygen species (ROS)7 ; MAPKs JNK2 and extracellular signal-regulated kinase 5 (ERK5)6,10 ; and the Rho/Rho-associated protein kinase (ROCK)–signaling module for cytoskeletal rearrangement.8 CD36 also desensitizes the inhibitory platelet protein kinase G (PKG) pathway,7 enhancing activation by classic agonists. Studies of CD36-null mice and CD36-deficient humans suggest that CD36 is not essential for normal hemostasis, but we and others hypothesized that it may potentiate prothrombotic activity under conditions when its ligands are greatly present, such as in dyslipidemia.2
Procoagulant platelets are a subpopulation of platelets generated during thrombosis.11,12 Exposure of anionic phospholipids, such as phosphatidylserine (PSer), on the platelet surface augments recruitment and activity of prothrombinase and tenase complexes.13 Procoagulant platelets are generated upon strong stimulation, which induces scramblase activation and PSer externalization in a process mediated by sustained elevation of cytoplasmic and mitochondrial calcium levels and cyclophilin D–dependent mitochondrial permeability transition pore (mPTP) formation.14-16 The pathways mediating procoagulant platelet formation have been thought of as distinct from the apoptotic pathways mediating platelet life span,17,18 and inhibition or elimination of apoptotic pathways and proteins, such as BH-3–mediated apoptosome formation and caspase activation, did not impact procoagulant platelet formation in response to strong agonists.18,19 Elevated platelet procoagulant activity has been reported in hypercholesterolemic individuals,20,21 but mechanisms underlying this are not clear.
Platelet CD36 signaling generates reactive oxygen species (ROS), which in turn activate the redox-sensitive MAPK ERK5.10 However, the signaling downstream of ERK5 remains incompletely defined. ERK5, by increasing expression of the Rho family GTPase Rac and the ribosomal s6 family kinase p70S6K, has been shown to promote maladaptive platelet signaling in the setting of myocardial infarction, and to distinguish the reported different roles for platelet activation in ST-segment elevation myocardial infarction (STEMI) compared with non-ST segment elevation.22,23 In addition, ERK5 is a critical downstream component of ristocetin-induced glycoprotein Ib-IX (GPIb-IX) activation through its enhancing effect on phosphatidylinositol 3-kinase (PI3K)/Akt signaling through casein kinase II.24 In nucleated cells, ERK5 promotes cell survival and proliferation and limits cell death by enhancing the transcription and expression of survival genes,25,26 but it is unclear whether ERK5 promotes the same mechanism in platelets.
Here, we report that oxLDL-CD36 signaling cross-talks with platelet GPVI signaling to enhance surface exposure of PSer. Surprisingly, unlike the intracellular processes previously implicated in strong agonist-induced procoagulant platelet formation, oxLDL-CD36–initiated PSer exposure requires apoptotic caspases and the downstream CD36 signaling effectors Src kinases, hydrogen peroxide, and ERK5. Activation of this CD36-initiated pathway by oxLDL in vitro or hyperlipidemia in vivo leads to enhanced fibrin formation and promotes arterial thrombosis.
Methods
Detecting PSer externalization
Washed human platelets were prestimulated with 50 μg/mL LDL or oxLDL up to 30 minutes at room temperature in the presence or absence of 1 μg/mL anti-CD36 FA6-152 or 1 μg/mL immunoglobulin G (IgG) control; 10 μM PP2 or PP3; 10 μM BIX02188, XMD8-92, or SP600125; 100 μM BAPTA-AM; 2000 U/mL polyethylene glycol (PEG)-catalase or denatured (boiled) PEG-catalase; 100 μM Z-VAD–FMK, or 5 μM cyclosporin A (CsA). For antibody inhibition studies, platelets were pretreated with 10 μg/mL IV.3 to prevent activation of platelets through FcγRIIa. Platelets were then stimulated up to 15 minutes with a low or high dose of convulxin (CVX; 50 ng/mL or 500 ng/mL), adenosine 5′-diphosphate (ADP; 1 μM or 10 μM), or thrombin (THR; 0.1 U/mL or 1.0 U/mL) followed by 15 minutes of staining with fluorophore-conjugated annexin V or lactadherin. Platelets were immediately fixed with 2% paraformaldehyde followed by flow cytometry analysis with an LSRII flow cytometer (BD Biosciences).
Fibrin formation assay
Fibrin formation was assayed as previously described.27,28 Washed human platelets were isolated from healthy donors and incubated with 50 μg/mL LDL, oxLDL, or phosphate-buffered saline (PBS) (1 hour, 37°C), followed by 250 ng/mL CVX, or a combination of 1.0 U/mL THR and 500 ng/mL CVX (7 minutes, 37°C). For some experiments, 7.5 μg/mL annexin V, 1 mM Gly-Pro-Arg-Pro (GPRP), 1 μg/mL IgG or FA6-152, 10 μM BIX02188 or XMD8-92, or 100 μM Z-VAD–FMK were added to platelets before stimulation with oxLDL. The treated, washed platelets (20 μL) were incubated with citrated plasma (20 μL; pooled from 3 separate donors), and fibrin formation was initiated with a mixture of 0.05 pM tissue factor and 5 mM CaCl2 (60 μL) and monitored at 405 nm for 20 minutes. Onset times, defined as the time to reach 5% of the peak, and peak absorbance were calculated using GraphPad Prism v.7.0d (GraphPad Software).
In vivo arterial thrombosis microscopy
Animals were placed on a standard chow or western high-fat diet for 6 to 10 weeks. Intravital imaging of platelet and fibrin accumulation in mouse cremaster arterioles was performed as previously described.29 After the mice were anesthetized, the cremaster muscle was exposed and superfused with warm saline during the experiment. Arteriolar wall injury was induced with a micropoint laser ablation system (Intelligent Imaging Innovations). Fluorescence images were captured using a high-speed camera (Orca Flash 4.0; Hamamatsu). Data were collected for at least 5 minutes following injury.
Study approval
Healthy human subjects gave written informed consent before standard phlebotomy. All studies with human samples were in accordance to the Medical College of Wisconsin’s Institutional Review Board. All animal studies were in accordance to the Institutional Animal Care and Use Committee of Medical College of Wisconsin and University of North Carolina.
Results
oxLDL promotes procoagulant PSer exposure
The impact of oxLDL on PSer exposure was investigated in washed human platelets using fluorophore-conjugated annexin V and lactadherin. In support of our hypothesis, oxLDL but not native LDL, led to binding of annexin V in a concentration-dependent manner, with 22% ± 3.5% of platelets showing fluorescence at 100 μg/mL oxLDL (Figure 1A-B). This concentration of oxLDL is within the reported range of saturable binding to platelet CD36.2 Kinetic analysis showed detectable binding of lactadherin at 1 minute with maximal binding between 5 and 15 minutes (Figure 1C). Because costimulation with both protease-activated receptor (PAR) and GPVI is required for maximal PSer-positive procoagulant platelets,30 we also examined whether oxLDL could enhance classic agonist-induced PSer exposure. To mimic physiologic conditions, platelets were sensitized with oxLDL before stimulating with a low or high dose of THR, ADP, or CVX. Consistent with previous reports, single-agonist stimulation with THR or ADP induced only minimal Pser externalization, whereas ∼20% of platelets became PSer-positive after CVX stimulation, similar to that seen with oxLDL (Figure 1D). oxLDL pretreatment significantly potentiated CVX (GPVI)-mediated PSer externalization, with >80% of platelets becoming PSer-positive. Interestingly, the potentiating effect of oxLDL was not seen in the context of ADP or low-dose THR stimulation, and was only modest in the context of high-dose THR (twofold; P = .04), suggesting cross-talk between the GPVI and CD36 pathways.
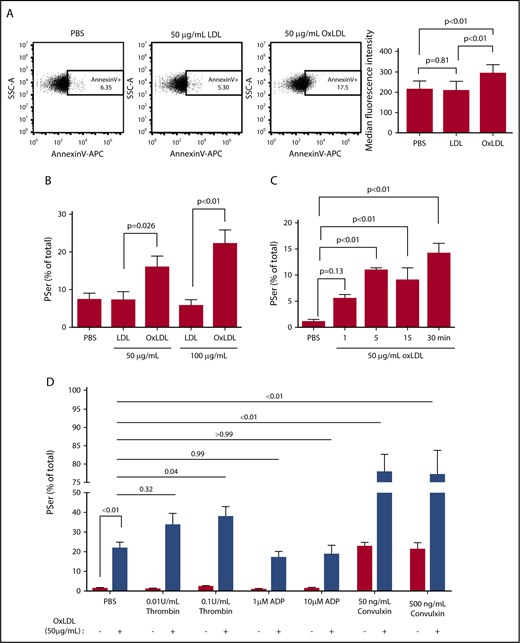
oxLDL promotes procoagulant PSer exposure. To detect exposed PSer, fluorophore-tagged annexin V or lactadherin binding to washed human platelets (1 × 108/mL) was measured by flow cytometry. Representative dot plots of annexin V binding to platelets stimulated with PBS, 50 μg/mL LDL or oxLDL for 15 minutes (A). Percent (%)-positive platelet staining with annexin V after exposure to PBS, 50 μg/mL or 100 μg/mL LDL or oxLDL (B), percent-positive staining with lactadherin up to 30 minutes with 50 μg/mL oxLDL (C) and oxLDL pretreatment followed by a low or high dose of classic agonists (D). Data represented as mean ± standard error of the mean (SEM). P value was determined by 1-way analysis of variance (ANOVA) with Tukey posthoc analysis in panel B or Dunnet post hoc analysis in panel C. P value was determined by 2-way ANOVA with Tukey post hoc analysis in panel D. N = 5 different donors in panel B, 3 different donors in panels A and C, and 4 different donors in panel D.
oxLDL promotes procoagulant PSer exposure. To detect exposed PSer, fluorophore-tagged annexin V or lactadherin binding to washed human platelets (1 × 108/mL) was measured by flow cytometry. Representative dot plots of annexin V binding to platelets stimulated with PBS, 50 μg/mL LDL or oxLDL for 15 minutes (A). Percent (%)-positive platelet staining with annexin V after exposure to PBS, 50 μg/mL or 100 μg/mL LDL or oxLDL (B), percent-positive staining with lactadherin up to 30 minutes with 50 μg/mL oxLDL (C) and oxLDL pretreatment followed by a low or high dose of classic agonists (D). Data represented as mean ± standard error of the mean (SEM). P value was determined by 1-way analysis of variance (ANOVA) with Tukey posthoc analysis in panel B or Dunnet post hoc analysis in panel C. P value was determined by 2-way ANOVA with Tukey post hoc analysis in panel D. N = 5 different donors in panel B, 3 different donors in panels A and C, and 4 different donors in panel D.
Rapid PSer externalization by oxLDL is mediated predominantly through a caspase pathway, not mPTP
A hallmark of classic procoagulant platelet formation is the THR/CVX-mediated cyclophilin D–sensitized mPTP formation,30 accompanied by loss of mitochondrial membrane potential (ΔΨm). This can be readily probed using tetramethylrhodamine methyl ester.31 Surprisingly, loss of ΔΨm was not observed by oxLDL. This is in marked contrast to the significant loss of ΔΨm observed in the classic THR/CVX-stimulated procoagulant platelets (supplemental Figure 1A). CsA, a peptidylprolyl isomerase inhibitor, inhibits cyclophilin D, a peptidylprolyl isomerase that links calcium flux to mPTP-mediated PSer externalization.32 CsA completely blocked THR/CVX-mediated PSer exposure and CVX-alone PSer exposure (Figure 2A; supplemental Figure 1B), but had no impact on oxLDL-mediated PSer exposure, strongly suggesting that mPTP formation is neither associated with nor required for oxLDL-mediated PSer externalization. Alternatively, the Bak/Bax-dependent apoptosome pathway is known to induce slow PSer externalization contributing to clearance of “aged” platelets.30 The pan caspase inhibitor Z-VAD–FMK had no impact on rapid PSer externalization induced by THR/CVX30,33,34 or CVX alone (supplemental Figure 1B), but surprisingly, PSer exposure induced by oxLDL was largely prevented (Figure 2A; P = .02), strongly suggesting a caspase-dependent mechanism. PSer externalization observed with oxLDL was not prevented by the caspase 3–specific inhibitor Z-DEVD–FMK (supplemental Figure 1D), indicating a role for other caspase members to mediate PSer externalization.
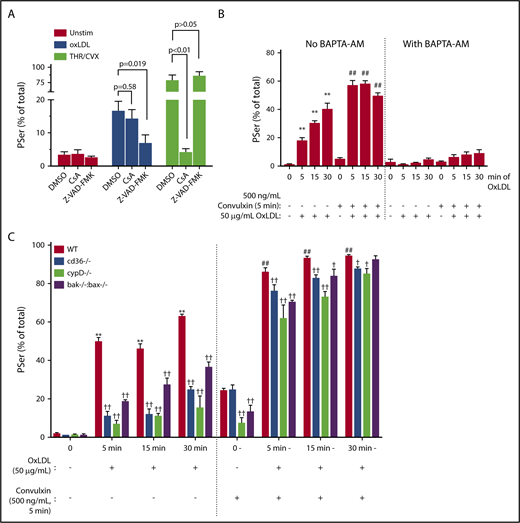
Rapid PSer externalization by oxLDL is by a calcium-dependent caspase pathway. (A) Human platelets (1 × 108/mL) were pretreated with 100 μM Z-VAD–FMK or 5 μM CsA followed by 15 minutes of stimulation with 50 μg/mL oxLDL or 7 minutes stimulation with 0.1 U/mL THR in the presence of 500 ng/mL CVX. (B) Human platelets (30 × 103/μL) were pretreated with 100 μM BAPTA-AM followed by oxLDL sensitization up to 30 minutes followed by 5 minutes of GPVI activation by 500 ng/mL CVX. (C) Gel-filtered platelets (30 × 103/μL) from WT, CD36-null, CypD-null, or Bak/Bax double-null mice were sensitized with 50 μg/mL oxLDL up to 30 minutes followed by 5 minutes of GPVI activation with 500 ng/mL CVX or buffer as a control. Percent-positive for annexin V binding was measured by flow cytometry. Data represented as mean ± SEM and analyzed by 1-way ANOVA with Tukey posthoc analysis. **P < .01 compared to WT no stimulation (no oxLDL, no CVX). ##P < .01 compared to WT with CVX alone (no oxLDL). †P < .05, ††P < .01, compared to their respective WT with oxLDL and CVX stimulation group. N of >3 different donors in panels A and B; platelets from 3 different age-matched mice per strain in panel C.
Rapid PSer externalization by oxLDL is by a calcium-dependent caspase pathway. (A) Human platelets (1 × 108/mL) were pretreated with 100 μM Z-VAD–FMK or 5 μM CsA followed by 15 minutes of stimulation with 50 μg/mL oxLDL or 7 minutes stimulation with 0.1 U/mL THR in the presence of 500 ng/mL CVX. (B) Human platelets (30 × 103/μL) were pretreated with 100 μM BAPTA-AM followed by oxLDL sensitization up to 30 minutes followed by 5 minutes of GPVI activation by 500 ng/mL CVX. (C) Gel-filtered platelets (30 × 103/μL) from WT, CD36-null, CypD-null, or Bak/Bax double-null mice were sensitized with 50 μg/mL oxLDL up to 30 minutes followed by 5 minutes of GPVI activation with 500 ng/mL CVX or buffer as a control. Percent-positive for annexin V binding was measured by flow cytometry. Data represented as mean ± SEM and analyzed by 1-way ANOVA with Tukey posthoc analysis. **P < .01 compared to WT no stimulation (no oxLDL, no CVX). ##P < .01 compared to WT with CVX alone (no oxLDL). †P < .05, ††P < .01, compared to their respective WT with oxLDL and CVX stimulation group. N of >3 different donors in panels A and B; platelets from 3 different age-matched mice per strain in panel C.
Next, we tested the role of intracellular calcium in oxLDL stimulation because calcium is a critical component to PSer externalization by classic agonists.35 In the absence of exogenous calcium, oxLDL induced PSer externalization from 18% ± 1.8% in 5 minutes up to 40% ± 3.8% in 30 minutes (Figure 2B). Platelet sensitization by oxLDL before activating with CVX induced further PSer externalization maximizing at 58% ± 1.9% within 5 to 15 minutes. Platelets were then treated with the calcium chelator BAPTA-AM followed by stimulation with either oxLDL alone or oxLDL with CVX. The presence of BAPTA-AM completely prevented PSer externalization by oxLDL either alone or in the presence of CVX, suggesting a critical role for intracellular calcium in PSer externalization by oxLDL.
We then studied platelets from wild-type (WT) C57Bl/6 mice or mice lacking CD36, cyclophilin D, or Bak/Bax to provide genetic confirmation of the human pharmacologic studies. As shown in Figure 2C, murine platelets stimulated with oxLDL displayed time-dependent increase in PSer, which was further substantially increased in the presence of CVX. oxLDL-induced PSer externalization alone was inhibited by >50% in CD36, CypD, and Bak/Bax-null platelets. PSer by oxLDL sensitization followed by CVX stimulation was partially inhibited in CD36, CypD, and Bak/Bax-null platelets.
oxLDL-mediated platelet PSer exposure requires CD36 signaling through the redox sensor MAPK ERK5
CD36 is the major high-affinity platelet receptor for oxidized lipids in oxLDL particles.2,36 In the presence of the CD36-blocking monoclonal antibody FA6-152, oxLDL-stimulated PSer exposure in human platelets was completely inhibited (Figure 3A), consistent with the results seen using CD36-null murine platelets.
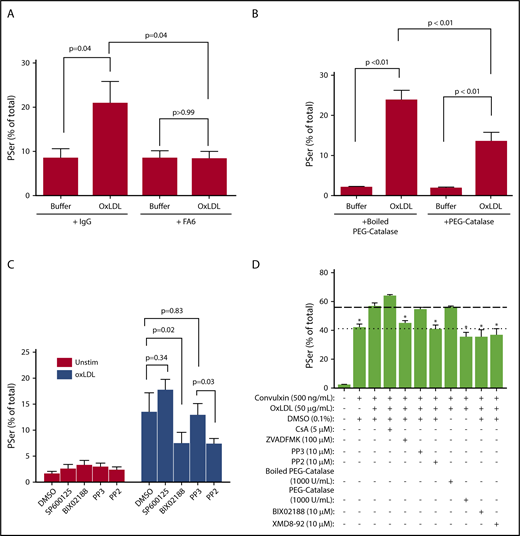
oxLDL-mediated platelet PSer exposure requires CD36 signaling through the redox sensor MAPK ERK5. Human platelets (1 × 108/mL) were pretreated with 1 μg/mL CD36 blocking antibody FA6-152 or nonimmunizing IgG isotype control (A), 2000 U/mL denatured boiled PEG catalase or native PEG catalase (B), 10 μM Src kinase inhibitor PP2, or control analog inhibitor PP3, 10 μM MEK5/ERK5 inhibitor BIX 02188 or JNK inhibitor SP600125 (C) or the indicated concentrations (D). Percent-positive annexin V binding was measured by flow cytometry after 50 μg/mL oxLDL only in panels A-C or 50 μg/mL oxLDL in the presence of 500 ng/mL CVX in panel D. Data represented as mean ± SEM and analyzed by 1-way ANOVA with Tukey post hoc analysis. *P < .05 compared to oxLDL with CVX in panel D. N of 4 different donors in panel A; 4 different donors in panel B; >3 donors in panel C; and 3 different donors in panel D.
oxLDL-mediated platelet PSer exposure requires CD36 signaling through the redox sensor MAPK ERK5. Human platelets (1 × 108/mL) were pretreated with 1 μg/mL CD36 blocking antibody FA6-152 or nonimmunizing IgG isotype control (A), 2000 U/mL denatured boiled PEG catalase or native PEG catalase (B), 10 μM Src kinase inhibitor PP2, or control analog inhibitor PP3, 10 μM MEK5/ERK5 inhibitor BIX 02188 or JNK inhibitor SP600125 (C) or the indicated concentrations (D). Percent-positive annexin V binding was measured by flow cytometry after 50 μg/mL oxLDL only in panels A-C or 50 μg/mL oxLDL in the presence of 500 ng/mL CVX in panel D. Data represented as mean ± SEM and analyzed by 1-way ANOVA with Tukey post hoc analysis. *P < .05 compared to oxLDL with CVX in panel D. N of 4 different donors in panel A; 4 different donors in panel B; >3 donors in panel C; and 3 different donors in panel D.
ROS, including superoxide radical anion and hydrogen peroxide are generated from NADPH oxidase downstream of platelet CD36 signaling.7,10 We previously showed that PEG catalase, an enzyme that degrades hydrogen peroxide, prevented platelet aggregation induced by oxLDL.10 As shown in Figure 3B, PEG catalase significantly decreased oxLDL-induced platelet PSer from 24% down to 14%. Inactivating the enzyme by denaturing had no impact on PSer exposure, suggesting that ROS induced by CD36 signaling plays a role in PSer externalization.
ROS activate redox-sensitive signaling pathways in platelets, including the MAPK ERK5.10 As shown in Figure 3C, the pharmacologic MEK5/ERK5 inhibitor BIX02188, which we previously showed to prevent oxLDL-induced platelet activation and aggregation,10 similarly inhibited PSer exposure by oxLDL. Another MAPK, JNK, can be activated in the settings of CD36 signaling6 and oxidant stress.37 To determine whether JNK has a role in promoting CD36-dependent PSer externalization by oxLDL, the pharmacologic inhibitor of JNK, SP600125, was used at concentrations previously demonstrated to inhibit CD36 signaling.6 Surprisingly, oxLDL-induced PSer exposure was not prevented. Furthermore, Src family kinases, particularly Fyn and Lyn, are recruited to platelet CD366 and have been proposed to primarily function upstream of both MAPKs ERK5 and JNK.6,10,24,38 Pretreating platelets with the broad-spectrum Src family kinase inhibitor PP239 decreased oxLDL-induced PSer exposure to the same extent as BIX02188, whereas the control analog PP3 inhibitor had no effect. These data suggest that ERK5, acted upon by upstream Src family kinases, is the major MAPK redox sensor for CD36 to promote PSer externalization.
Because we observed potentiation of surface PSer exposure by stimulating platelets with oxLDL before adding CVX, we hypothesized that caspases, cyclophilin D, Src family kinases, hydrogen peroxide, and ERK5 are functionally important in the crosstalk between CD36 and GPVI. Therefore, we pretreated platelets with the inhibitors to these mediators before stimulating with oxLDL and CVX. As shown in Figure 3D and supplemental Figure 1E, pretreatment with Z-VAD–FMK, PP2, PEG catalase, and the 2 ERK5 inhibitors BIX02188 and XMD8-92 prevented the incremental PSer exposure induced by oxLDL prior to CVX stimulation. As expected, pretreatment with CsA, PP3, and boiled PEG catalase had no impact.
CD36/ERK5 signaling is a key driver of platelet caspase activation by oxLDL
Next, we investigated the function of the CD36/ROS/ERK5 pathway in mediating caspase activation in response to oxLDL. Caspase activity, quantified as the ratio of cleaved caspase 3 to procaspase 3, was elevated in oxLDL-treated platelets comparable to that seen in platelets treated with the BH-3–mimetic ABT-737 (Figure 4A). Caspase activation was also confirmed using a colorimetric assay for caspase 3 cleavage of the substrate DEVD (Figure 4B), which showed that oxLDL indeed induced caspase activity and ABT-737 stimulated 20% more activity than oxLDL. We postulated that CD36 and ERK5 are upstream activators of caspases by oxLDL. Pretreatment of platelets with the CD36-blocking antibody FA6-152, but not control IgG, prevented oxLDL-induced caspase 3 cleavage (Figure 4C). Similarly, the MEK5/ERK5 inhibitor BIX02188 abrogated caspase 3 cleavage (Figure 4D). These data indicate that oxLDL promotes caspase activation through platelet CD36 and ERK5.
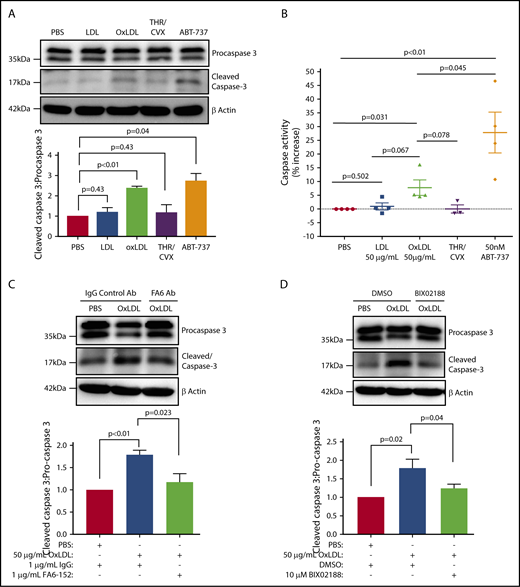
Platelet CD36/ERK5 are key drivers of caspase activation by oxidized lipids. (A) Cleaved caspase 3 was detected by immunoblot as a marker of caspase activity after human platelets were stimulated with buffer, 50 μg/mL LDL or oxLDL, and 50 nM ABT-737 for 60 minutes; 0.1 U/mL THR with 50 ng/mL CVX for 7 minutes were used as a negative control. (B) Human platelets were treated according to panel A. The whole-cell lysate from each treatment was incubated with the caspase 3 substrate DEVD-pNa according to the manufacturer’s protocol. The slopes of p-nitroaniline absorbance were used to indicate activity. FA6-152 or an IgG control antibody (1 μg/mL) was used to determine CD36 dependency (C) and 10 μM BIX02188 was used to determine ERK5 dependency (D). Densitometric analysis of the ratio of cleaved caspase 3/procaspase 3 was indicated with each blot. Blots are representative of at least 3 separate experiments from 3 different donors in panels A-D. Data represented as mean ± SEM.
Platelet CD36/ERK5 are key drivers of caspase activation by oxidized lipids. (A) Cleaved caspase 3 was detected by immunoblot as a marker of caspase activity after human platelets were stimulated with buffer, 50 μg/mL LDL or oxLDL, and 50 nM ABT-737 for 60 minutes; 0.1 U/mL THR with 50 ng/mL CVX for 7 minutes were used as a negative control. (B) Human platelets were treated according to panel A. The whole-cell lysate from each treatment was incubated with the caspase 3 substrate DEVD-pNa according to the manufacturer’s protocol. The slopes of p-nitroaniline absorbance were used to indicate activity. FA6-152 or an IgG control antibody (1 μg/mL) was used to determine CD36 dependency (C) and 10 μM BIX02188 was used to determine ERK5 dependency (D). Densitometric analysis of the ratio of cleaved caspase 3/procaspase 3 was indicated with each blot. Blots are representative of at least 3 separate experiments from 3 different donors in panels A-D. Data represented as mean ± SEM.
oxLDL-mediated fibrin formation ex vivo is prevented by inhibiting CD36, ERK5, and apoptotic caspases
To assess the functional importance of oxLDL-mediated procoagulant platelet formation, the ability to facilitate tissue factor (TF)-induced fibrin formation was investigated. Fibrin formation in platelet-rich plasma can be readily detected by light absorbance at 405 nm after addition of Ca2+ and picomolar levels of TF.28 This assay demonstrated rapid initiation and potentiation of fibrin formation by “classic” costimulation with THR and CVX (Figure 5A; supplemental Table 1). PSer dependence was shown by inhibition of fibrin formation by annexin V, which masks exposed PSer. The peptide GPRP, which blocks fibrin polymerization, was used to validate that the change in absorbance was due to fibrin polymer formation.
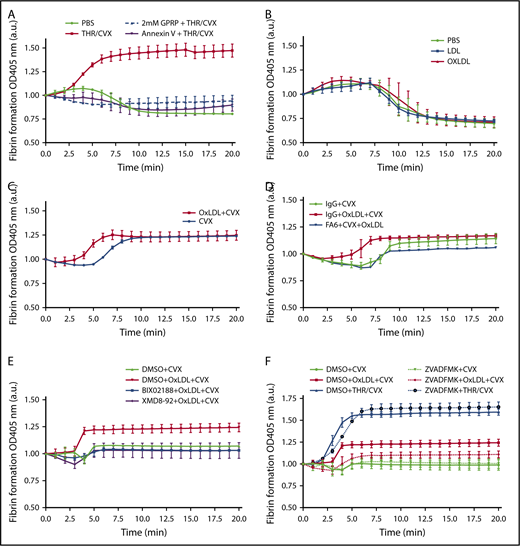
Fibrin formation ex vivo by oxLDL is prevented by inhibiting CD36, ERK5, and apoptotic caspases. (A) Human platelets (3 × 108/mL) were pretreated with the fibrin polymerization inhibitor GPRP or PSer inhibitor annexin V before stimulating with 0.1 U/mL THR with 500 ng/mL CVX. (B) Platelets were stimulated with PBS, 50 μg/mL LDL or oxLDL alone. (C) Platelets were sensitized with 50 μg/mL oxLDL followed by 250 ng/mL CVX stimulation or stimulated with 250 ng/mL CVX alone. Platelets were treated with the 1 µg/mL CD36-blocking FA6 or IgG antibody (D), DMSO (0.5%), 10 µM MEK5/ERK5 inhibitor BIX02188 or XMD8-92 (E), or with 100 µM caspase inhibitor Z-VAD–FMK (F) before stimulating with 250 ng/mL CVX alone or 50 μg/mL oxLDL with CVX; 0.1 U/mL THR with 500 ng/mL CVX stimulation was used as a control for Z-VAD–FMK treatment in panel F. Fibrin polymerization was measured at 405 nm absorbance over time with at least 3 separate donors per stimulation. Data represented as mean ± SEM. N of >3 different donors in panels A-F.
Fibrin formation ex vivo by oxLDL is prevented by inhibiting CD36, ERK5, and apoptotic caspases. (A) Human platelets (3 × 108/mL) were pretreated with the fibrin polymerization inhibitor GPRP or PSer inhibitor annexin V before stimulating with 0.1 U/mL THR with 500 ng/mL CVX. (B) Platelets were stimulated with PBS, 50 μg/mL LDL or oxLDL alone. (C) Platelets were sensitized with 50 μg/mL oxLDL followed by 250 ng/mL CVX stimulation or stimulated with 250 ng/mL CVX alone. Platelets were treated with the 1 µg/mL CD36-blocking FA6 or IgG antibody (D), DMSO (0.5%), 10 µM MEK5/ERK5 inhibitor BIX02188 or XMD8-92 (E), or with 100 µM caspase inhibitor Z-VAD–FMK (F) before stimulating with 250 ng/mL CVX alone or 50 μg/mL oxLDL with CVX; 0.1 U/mL THR with 500 ng/mL CVX stimulation was used as a control for Z-VAD–FMK treatment in panel F. Fibrin polymerization was measured at 405 nm absorbance over time with at least 3 separate donors per stimulation. Data represented as mean ± SEM. N of >3 different donors in panels A-F.
Surprisingly, despite the presence of externalized PSer on the platelet surface, oxLDL treatment alone did not potentiate TF-induced fibrin formation. The amount and rate of fibrin formation was similar in oxLDL, LDL, and buffer-stimulated platelets (Figure 5B; supplemental Table 1). oxLDL, however, significantly amplified the effect of CVX as shown in Figure 5C and supplemental Table 1; oxLDL pretreatment decreased CVX-stimulated lag time for fibrin formation from 5.6 ± 0.06 minutes to 3.4 ± 0.1 minutes. Pretreatment of platelets with the CD36 inhibitory antibody FA6-152 abrogated oxLDL-sensitized CVX-initiated fibrin formation (Figure 5D; supplemental Table 1). Similarly, treatment with the MEK5/ERK5 inhibitor BIX02188 or ERK5 inhibitor XMD8-92 also abrogated oxLDL-enhanced lag time and peak fibrin formation induced by CVX (Figure 5E; supplemental Table 1). Finally, the relevance of caspase activity to the oxLDL-enhanced fibrin formation by platelets was tested by pretreating platelets with the caspase inhibitor Z-VAD–FMK. Although caspase inhibition had no effect on fibrin formation in THR/CVX-stimulated platelets, consistent with previous reports,18,19 treating platelets with Z-VAD–FMK largely abrogated the increase in peak fibrin formation induced by oxLDL and CVX (Figure 5F; supplemental Table 1).
Hyperlipidemic apoE-null mice have increased fibrin accumulation in vivo after vascular injury and the phenotype is rescued by CD36 or platelet ERK5 deletion
Our previous studies demonstrated the importance of platelet CD36 in the development of atherothrombosis in mice. In FeCl3-induced carotid artery thrombosis, absence of CD36 eliminated the accelerated thrombotic occlusion observed in animals on a high-fat diet.2,5 Fibrin formation was not tested initially in this model. Thus, we more closely examined the role of platelet-mediated fibrin formation in diet-induced thrombosis. Using a laser-induced cremasteric artery injury model, as shown in Figure 6A-C and supplemental Videos 1 and 2, apoE-null mice fed a high-fat diet demonstrated both increased platelet and fibrin accumulation compared with chow-fed animals. In the high-fat-diet–fed animals, a procoagulant phenotype with elevated fibrin and platelet accumulation was observed within 30 seconds of laser injury (Figure 6A and B-C left panel) and the thrombus occasionally occluded the vessel without resolving. In control chow-diet–fed mice, the laser-induced thrombi resolved over time with no vessel occlusion observed (Figure 6A and B-C right panels; supplemental Video 3). The role of CD36-mediated signaling was investigated using apoE:CD36 double-null mice.2 On the high-fat diet, these mice showed similar platelet and fibrin accumulation as control-diet–fed animals (Figure 6A-C; supplemental Videos 2-4).
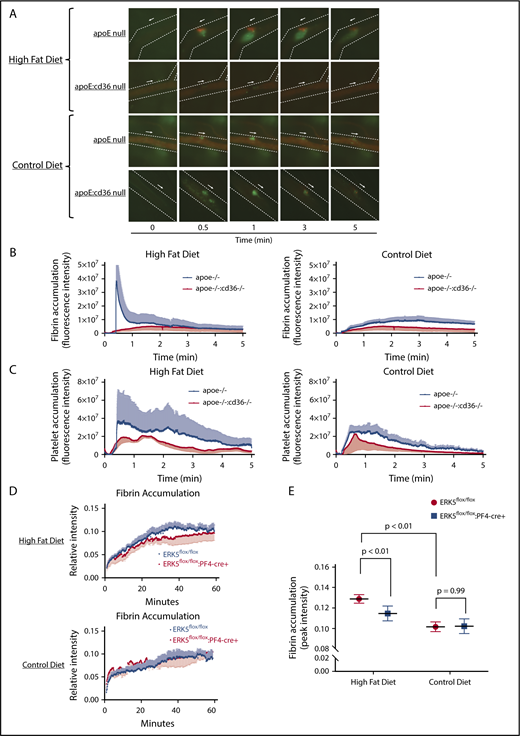
Hyperlipidemic apoE-null animals with CD36 or ERK5 deficiency showed decreased fibrin accumulation in vivo. ApoE-null or apoE:CD36 double-null mice at 8 weeks of age were fed a control or high-fat diet for at least 6 weeks before performing laser-induced in vivo arterial thrombosis on the cremasteric artery (A-C). Video microscopy of the platelet accumulation (in green) and fibrin accumulation (in red) in real time up to 5 minutes is shown in panel A; the arrow indicates the direction of arterial flow and the side the arrow is on indicates the side of vessel injury. Quantification of fibrin (B) and quantification of platelets (C). ApoE-null mice at 8 weeks of age were irradiated and were transplanted with bone marrow from ERK5flox/flox or ERK5flox/flox:PF4-cre+ mice. (D-E) After 2 weeks of recovery, the apoE-null chimeric mice were put on chow or a high-fat diet for at least 6 weeks before performing the adventitial tissue-mediated in vivo arterial thrombosis on the carotid artery (supplemental Figure 2). The quantification of peak fibrin accumulation with this model is shown in panel E by using the 1-site nonlinear curve fitting model. Video microscopy images of fibrin accumulation in real time are shown in supplemental Figure 2. N of >5 age-matched male mice per group in panels A-C; >6 age-matched male or female mice per group in panels D-E.
Hyperlipidemic apoE-null animals with CD36 or ERK5 deficiency showed decreased fibrin accumulation in vivo. ApoE-null or apoE:CD36 double-null mice at 8 weeks of age were fed a control or high-fat diet for at least 6 weeks before performing laser-induced in vivo arterial thrombosis on the cremasteric artery (A-C). Video microscopy of the platelet accumulation (in green) and fibrin accumulation (in red) in real time up to 5 minutes is shown in panel A; the arrow indicates the direction of arterial flow and the side the arrow is on indicates the side of vessel injury. Quantification of fibrin (B) and quantification of platelets (C). ApoE-null mice at 8 weeks of age were irradiated and were transplanted with bone marrow from ERK5flox/flox or ERK5flox/flox:PF4-cre+ mice. (D-E) After 2 weeks of recovery, the apoE-null chimeric mice were put on chow or a high-fat diet for at least 6 weeks before performing the adventitial tissue-mediated in vivo arterial thrombosis on the carotid artery (supplemental Figure 2). The quantification of peak fibrin accumulation with this model is shown in panel E by using the 1-site nonlinear curve fitting model. Video microscopy images of fibrin accumulation in real time are shown in supplemental Figure 2. N of >5 age-matched male mice per group in panels A-C; >6 age-matched male or female mice per group in panels D-E.
We previously reported that ERK5 is essential in promoting platelet accumulation in hyperlipidemic conditions using an adventitial tissue-mediated carotid artery thrombosis model.10 This model promotes thrombosis by exposing the thrombogenic surface of the adventitial tissue of the epigastric artery to arterial flow in the carotid artery by transplantation,40 and, importantly, is not mediated by oxidative damage to the vessel wall. Using this model, we now demonstrate that irradiated apoE-null mice transplanted with bone marrow from ERK5-expressing (ERK5flox/flox) mice and then fed a high-fat diet for 6 weeks showed enhanced peak fibrin accumulation (Figure 6D top and E left; supplemental Figure 2). Transplant of platelet-specific ERK5-deficient bone marrow (ERK5flox/flox:PF4-cre+), however, into irradiated apoE-null mice fed a high-fat diet did not show enhanced fibrin accumulation (Figure 6D bottom and E right; supplemental Figure 2) and had similar levels of fibrin accumulation as chow-diet–fed mice. Fibrin formation, however, ultimately reached the same peak with either ERK5 replete or deficient platelets, reflecting the rapid dynamics of fibrin formation and breakdown in this model.
Discussion
Atherothrombosis is a major complication of dyslipidemia and can lead to myocardial infarction and stroke. Platelet activation and fibrin formation play essential roles in atherothrombosis, but underlying mechanisms that promote these processes remain incompletely defined. As modeled in Figure 7, we showed that ERK5 links platelet CD36 to a procoagulant phenotype induced by oxLDL. This mechanism is mediated by direct activation of the ERK5 pathway by CD36 requiring Src family kinases and hydrogen peroxide. ERK5 then promotes cellular caspase activation leading to procoagulant PSer externalization. Furthermore, crosstalk between the CD36 and GPVI pathways potentiates PSer externalization requiring Src kinases, ERK5, and intracellular calcium. CD36/ERK5 signaling promotes fibrin formation and accumulation, as shown by 2 models of arterial thrombosis. Although the procoagulant phenotype mediated by the cyclophilin D–mPTP pathway is a well-described pathway to promote TF-mediated thrombosis and hemostasis,14,30 this is the first report of a rapid PSer externalization mechanism mediated by an alternative oxLDL-initiated caspase-dependent pathway that is relevant in the pathophysiology of arterial thrombosis.
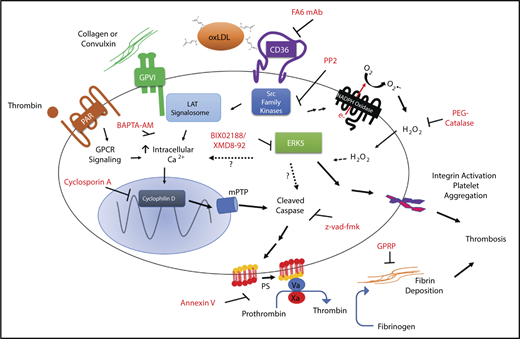
Model for the procoagulant properties of platelet CD36/ERK5 signaling. In dyslipidemic conditions, circulating oxidized lipids present in oxLDL particles are recognized by platelet CD36. This promotes immediate recruitment of Src family kinases and generation of ROS by NADPH oxidase. Activation of MAPK ERK5 signaling by superoxide anion, and hydrogen peroxide promotes the apoptosome formation and caspase activity. The mechanisms of activating ERK5 by ROS are unclear. Caspases activate known scramblases present on the surface of the membrane and thus promote externalization of procoagulant phosphatidylserine. ERK5 also induces a signaling pathway that promotes integrin activation to enhance thrombosis. There is crosstalk between CD36 and GPVI downstream signaling, potentially through Src family kinases, ERK5, or calcium. This crosstalk enhances the externalization of phosphatidylserine through maladaptive apoptosome formation that is not common to the cyclophilin D/mPTP pathway induced by the strong agonists THR and CVX or the physiologic cell death pathway. Assembly of the prothrombinase and tenase complex to the anionic phospholipid enhances fibrin accumulation in vivo to support arterial thrombosis in dyslipidemic conditions.
Model for the procoagulant properties of platelet CD36/ERK5 signaling. In dyslipidemic conditions, circulating oxidized lipids present in oxLDL particles are recognized by platelet CD36. This promotes immediate recruitment of Src family kinases and generation of ROS by NADPH oxidase. Activation of MAPK ERK5 signaling by superoxide anion, and hydrogen peroxide promotes the apoptosome formation and caspase activity. The mechanisms of activating ERK5 by ROS are unclear. Caspases activate known scramblases present on the surface of the membrane and thus promote externalization of procoagulant phosphatidylserine. ERK5 also induces a signaling pathway that promotes integrin activation to enhance thrombosis. There is crosstalk between CD36 and GPVI downstream signaling, potentially through Src family kinases, ERK5, or calcium. This crosstalk enhances the externalization of phosphatidylserine through maladaptive apoptosome formation that is not common to the cyclophilin D/mPTP pathway induced by the strong agonists THR and CVX or the physiologic cell death pathway. Assembly of the prothrombinase and tenase complex to the anionic phospholipid enhances fibrin accumulation in vivo to support arterial thrombosis in dyslipidemic conditions.
The PSer externalization mediated by caspases and the apoptosome is known to be important in the physiologic processes of platelet cell death and clearance.41 This can be inhibited pharmacologically or through genetic deletion of Bak/Bax.18 Caspases promote cell death and PSer externalization by cleaving and activating the scramblase Xk–related protein 8 on the membrane.42 Bak/Bax has primarily been thought to function as a regulator of platelet clearance through a yet-to-be-defined mechanism.34,41 Bak/Bax-initiated caspase function in mice are both dispensable for platelet production and hemostasis. In caspase-9-null mice, homeostatic platelet clearance was unaffected, and the only procoagulant defect identified was a marked abrogation of pharmacologically-initiated (BcL-xL inhibitor ABT-737) and caspase-dependent PSer externalization.34 Thus, the relevance of caspase activation in thrombosis and hemostasis is unclear.
In this article, we report a pathophysiologic role of caspase-mediated platelet procoagulant activity in the context of dyslipidemia. Caspase activation in platelets requires CD36 to recognize oxLDL and activate ERK5. Caspases in turn promote procoagulant PSer externalization. Although the direct mechanism for ERK5 to induce the apoptosome is unclear and under investigation, we propose ERK5 as a mediator for CD36 to trigger maladaptive apoptosome formation in platelets. Genetic interruption of apoptosome formation using Bak/Bax double-null mice showed abrogation of PSer externalization by oxLDL (Figure 2C). Surprisingly, CypD-null mice also showed inhibition of PSer externalization by oxLDL alone or by sensitization of the CVX-induced GPVI activation. This phenotype was not observed by preventing CypD activation using the peptidylprolyl isomerase inhibitor cyclosporin A (Figure 2A), but only with the pan caspase inhibitor Z-VAD–FMK. The specific caspase member mediating PSer externalization by oxLDL is not defined, but likely involves multiple caspases because the caspase 3–specific inhibitor Z-DEVD–FMK showed limited inhibition of PSer (supplemental Figure 1D). Although we do not discount a potential role for CypD, we suggest caspases as the major pathway for oxLDL-induced PSer externalization.
Several reports indicated that oxLDL activation of CD36 promotes signaling components common to GPVI. These components include activation of Fyn and Syk, which are critical for signal transduction from the GPVI/FcRγ chain to the Lat signalosome.43 In addition, Vavs are key signaling adaptors within the LAT signalosome and are phosphorylated downstream of CD36 through a Src family kinase-dependent mechanism.9 Sensitization of the CVX/GPVI-induced PSer externalization by oxLDL may be related mechanistically to activation of components of the GPVI pathway, including a role for Src family kinases, ERK5, and intracellular calcium (Figures 2B and 3C-D). It is expected that coordinated activation of 1 or more members of the Src family kinases by both a GPVI agonist and CD36 ligand will potentiate platelet activation and subsequent PSer externalization. The surprising finding that the pan Src family kinase inhibitor prevented sensitization of the GPVI pathway (oxLDL with CVX) could be mechanistically linked to the Src family members involved (Figure 3D). However, the specific member involved is unclear because the pan Src family kinase inhibitor PP2 only partially prevented PSer externalization by GPVI (supplemental Figure 1C).
Generation of ROS is characteristic of oxidant stress during hyperlipidemia.7,10 THR and CVX are potent activators of ROS pathways in platelets30 with clinical evidence suggesting that PAR1 antagonists, such as vorapaxar, are efficacious in preventing recurrent atherothrombosis in patients with known coronary artery disease.44 Furthermore, differential roles for classic agonists in ERK5 activation, such as with THR/PAR1,22 were shown to be dependent on ROS.45 Despite a role for ROS in platelet activation, H2O2 degradation by catalase only partially prevented oxLDL-induced PSer externalization (shown in Figure 3B), which predictably corresponded with catalase partially preventing platelet aggregation by oxLDL.10 These data suggest that signaling mechanisms independent of ROS are important for PSer externalization by oxLDL.
Molecular events driving fibrin formation ex vivo and in vivo are initiated by recruitment of the prothrombinase and tenase complex.13 Spectrophotometric analysis of fibrin formation is sensitive and quantitative ex vivo.27,28 Interestingly, oxLDL sensitization of the GPVI pathway (oxLDL with CVX) induces an earlier onset time for fibrin formation that is CD36, ERK5, and caspase dependent (Figure 5). The difference in CVX-induced fibrin formation in Figure 5C-F may be due to the solvent used. Dimethyl sulfoxide (DMSO) could inhibit several aspects of platelet activation, so it is not surprising that it inhibits platelet procoagulant activity. Furthermore, these experiments were performed with equal platelet numbers between treatment, and the accelerated fibrin formation ex vivo by oxidized lipids supported our hypothesis. We complimented these studies with diet-induced murine models of thrombosis (Figure 6), which showed that diet-induced dyslipidemia promoted fibrin and platelet accumulation in vivo, but was rescued when CD36 and platelet ERK5 are absent. However, the enhanced fibrin accumulation in vivo could potentially be correlated with the levels of platelet accumulation, which could not be distinguished in the thrombosis models used and requires further analysis.
Together, these data suggest that activating caspases by CD36 signaling could be targeted to reduce thrombosis risk in conditions where CD36 ligands are greatly generated, such as in hyperlipidemia,2 diabetes mellitus,46 and chronic inflammation,5 without impacting normal hemostasis.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Rong Gloria Yuan and Rae Janecke for technical assistance, as well as the Thrombosis Core of the Blood Research Institute of BloodCenter of Wisconsin for assistance with the laser thrombosis model.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants 5R01HL111614 and 3R01HL111614-S1 (R.L.S.), R01 HL124018 and R01 HL141106-01A1 (C.N.M.), K08HL128856 and HL120200 (S.J.C.), and HL129193 (J.P.W.).
M.Y. was a PhD candidate at the Medical College of Wisconsin and this work was submitted in partial fulfillment for the degree.
Authorship
Contribution: M.Y. designed and performed the experiments and wrote the manuscript; A.K. designed and performed the experiments; M.L.S. performed the laser-induced in vivo cremaster arterial thrombosis model and wrote parts of the manuscript; B.C.C. performed the adventitial tissue-mediated in vivo carotid artery thrombosis model; N.O.S. performed experiments; J.P.W. designed experiments; S.J.C. and C.N.M. provided valuable input on the data and provided reagents; S.M.J. designed experiments and provided reagents; R.L.S. supervised the project and wrote the manuscript; and all of the authors analyzed the data and edited, and wrote parts of, the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Roy L. Silverstein, Hub for Medical Collaboration, Medical College of Wisconsin, Room 8745, 8701 West Watertown Plank Rd, Milwaukee, WI 53226; e-mail: rsilverstein@mcw.edu.

