Key Points
The MTD of AMG 232 was not reached. Dose escalation was discontinued due to gastrointestinal AEs at higher doses.
Evidence of clinical activity by AMG-232 was observed in some patients. Further evaluation is warranted.

Abstract
This open-label, phase 1 study evaluated the safety, pharmacokinetics, and maximum tolerated dose of AMG 232, an investigational oral, selective mouse double minute 2 homolog inhibitor in relapsed/refractory acute myeloid leukemia (AML). AMG 232 was administered orally once daily for 7 days every 2 weeks (7 on/off) at 60, 120, 240, 360, 480, or 960 mg as monotherapy (arm 1) or at 60 mg with trametinib 2 mg (arm 2). Dose-limiting toxicities (DLTs), adverse events (AEs), pharmacokinetics, clinical and pharmacodynamic response, and expression of p53 target genes were assessed. All 36 patients received AMG 232. No DLTs occurred in arm 1, and 360 mg was the highest test dose; dose escalation was halted due to gastrointestinal AEs at higher doses. One of ten patients in arm 2 had a DLT (grade 3 fatigue); 60 mg was the highest dose tested with trametinib. Common treatment-related AEs (any grade) included nausea (58%), diarrhea (56%), vomiting (33%), and decreased appetite (25%). AMG 232 exhibited linear pharmacokinetics unaffected by coadministration with trametinib. Serum macrophage inhibitor cytokine-1 and bone marrow expression of BAX, PUMA, P21, and MDM2 increased during treatment. Of 30 evaluable patients, 1 achieved complete remission, 4 had morphologic leukemia-free state, and 1 had partial remission. Four of 13 (31%) TP53-wild-type patients and 0 of 3 (0%) TP53-mutant patients were responders. AMG 232 was associated with gastrointestinal AEs at higher doses but had acceptable pharmacokinetics, on-target effects, and promising clinical activity warranting further investigation in patients with relapsed/refractory AML. This trial was registered at www.clinicaltrials.gov as #NCT02016729.
Introduction
The tumor suppressor p53 is a transcription factor encoded by the TP53 gene that is essential for cell cycle arrest and apoptosis of cancer cells.1,2 Mouse double minute 2 homolog (MDM2; known as HDM2 in humans) binds and inhibits the NH2 terminal transactivation domain of p53, blocking its transcription and causing its ubiquitination and degradation.3 MDM2 has become an attractive therapeutic target in the treatment of p53 wild-type (P53WT) cancers. Several MDM2 inhibitors are under investigation in clinical trials for the treatment of solid tumors and hematologic malignancies, including acute myeloid leukemia (AML).4,5
AMG 232 is an investigational oral, selective MDM2 inhibitor that restores p53 tumor suppression by blocking the MDM2-p53 interaction.6 In the phase 1 first-in-human study, AMG 232 had an acceptable tolerability and pharmacokinetic profile when administered up to the maximum tolerated dose (MTD) of 240 mg once daily for 7 days in a 21-day cycle in patients with P53WT advanced solid tumors or multiple myeloma.7
Preclinical studies have suggested that MDM2 inhibition synergizes with MEK inhibition against P53WT cells, including AML cells, and that the activity may be dependent on the proapoptotic proteins Puma (p53 upregulated modulator of apoptosis) and Bim (Bcl-2 interacting mediator of cell death).8-13 In RKO tumor xenograft models, AMG 232 had antitumor activity as monotherapy that was enhanced in combination with a MEK inhibitor.10 Furthermore, phase 1 clinical studies have shown evidence of efficacy with MDM2 inhibitors and MEK inhibitors in AML,14,15 suggesting that combination therapy may result in greater clinical activity. In clinical studies, increased blood levels of macrophage inhibitor cytokine-1 (MIC-1) has been used as a pharmacodynamic marker of treatment with other MDM2 inhibitors in patients with relapsed/refractory AML and in patients with other solid tumors, indicating on-target biological activity.15-20 Expression of the p53 target genes BAX, PUMA, P21, and MDM2 in leukemic bone marrow has also been demonstrated following treatment with MDM2 inhibitors.15-17
Trametinib is a MEK inhibitor indicated as monotherapy for unresectable or metastatic melanoma with BRAF V600E or V600K mutations or in combination therapy for the treatment of unresectable or metastatic melanoma with BRAF V600E or V600K mutations, metastatic non–small-cell lung cancer with BRAF V600E mutation, or locally advanced or metastatic anaplastic thyroid cancer with BRAF V600E mutation and no locoregional treatment option.21 This study assessed the safety and tolerability, pharmacokinetics, and MTD of AMG 232 as monotherapy or combined with trametinib in patients with relapsed/refractory AML.
Methods
Patients
Patients aged ≥18 years with pathologically documented, treatment-refractory or relapsed AML, Eastern Cooperative Oncology Group performance status ≤2, life expectancy >3 months, and adequate renal (serum creatinine <2.0 mg/dL or estimated glomerular filtration rate >40 mL/min/1.73m2), hepatic (aspartate aminotransferase and alanine aminotransferase ≤3.0× upper limit of normal [ULN], alkaline phosphatase <2.0× ULN, and bilirubin ≤1.5× ULN), and cardiac (left ventricular ejection fraction of at least the lower limit of normal) function were eligible for the study. Patients with 17p deletion based on cytogenetics or with TP53-mutant (P53MT) AML when TP53 mutational status was known were excluded from the study. Patients with complex karyotype (defined as AML exhibiting >3 cytogenetic abnormalities in bone marrow, not including inv(16), t(16;16), t(8;21), t(15;17), and t(9;11)) were excluded, as AMLs with complex karyotypes have high rates of TP53 mutation.22-25 Patients with complex karyotype with known P53WT status were allowed. Other exclusion criteria included acute promyelocytic leukemia or active central nervous system leukemia; history of interstitial lung disease, pneumonitis (arm 2); history or risk of retinal vein occlusion (arm 2 only); allogeneic stem cell transplantation within 8 weeks before study entry; ongoing immunosuppressive therapy or graft-versus-host disease; immune modulators or corticosteroids within 2 weeks before study entry; unresolved toxicities from prior anticancer therapy, excluding alopecia; antitumor therapy within 14 days before study entry; or prior treatment with an MDM2 inhibitor (arms 1 and 2) or MEK inhibitor (arm 2). Institutional review board approval was obtained for all study procedures. All patients provided informed consent before enrollment.
Study design and treatment
This open-label phase 1 study was conducted at 5 centers (www.clinicaltrials.gov #NCT02016729). The study was designed to investigate the safety and tolerability, MTD, pharmacokinetics, pharmacodynamics, and preliminary antitumor activity of AMG 232 as monotherapy or combined with trametinib in patients with relapsed/refractory AML. In the dose escalation, multiple-patient cohorts (3 or 4 patients each) were enrolled sequentially to receive AMG 232 orally once daily for 7 days every 2 weeks (7 days on, 7 days off) at the prespecified doses of 60, 120, 240, 480, and 960 mg as monotherapy (arm 1) or combined with trametinib 2 mg administered orally once daily (arm 2) until clinical progression, intolerability, or withdrawal of consent. The dose of AMG 232 in arm 2 was to be selected based on observations in arm 1. Enrollment in arms 1 and 2 was conducted in parallel. Intermediate doses (at a 1.5-fold increment) were allowed as needed for toxicity.
The dose-limiting toxicity (DLT) evaluation window was 28 days (2 cycles). DLTs were defined as any grade 3 or 4 treatment-related nonhematologic adverse event (AE) per Common Terminology Criteria for Adverse Events, version 4.0, where a relationship to AMG 232 cannot be ruled out, except for infections and grade 3 laboratory abnormalities without clinical significance. DLTs also included grade ≥3 nausea, vomiting, or diarrhea lasting >48 hours after management; grade 3 fatigue lasting >7 days; any treatment-related AEs not returning to grade ≤1 or baseline severity after a treatment delay up to 7 days; and pancytopenia in the presence of hypocellular bone marrow lasting >42 days. The MTD was estimated using a Bayesian logistic regression model using all DLT-evaluable patients. Dose escalation was considered complete if any of the following occurred: the first dose level ≥2 patients had a DLT in cycles 1 or 2; highest planned dose was met with no DLTs in cycles 1 or 2 at any dose level; the Bayesian logistic regression model model recommended the same dose >3 times; or 40 DLT-evaluable patients were enrolled. Treatment continued until disease progression, intolerable toxicity, or withdrawal of consent. At least 28 days of safety follow-up after the last dose was required to capture AEs before the protocol allowed dosing of patients to the next dose level. Dose escalation did not occur until after a dose-level review meeting was held. The meeting, per protocol, could not occur until all patients in a cohort were followed for a minimum of 28 days.
Study assessments
Safety
AEs (graded per Common Terminology Criteria for Adverse Events, version 4.0) were recorded for all enrolled patients.
Pharmacokinetic analysis
Plasma concentrations of AMG 232 and its glucuronide metabolite were determined by validated assay of liquid chromatography (LC) with tandem mass spectrometric detection (MS/MS) using calibration curves with the range 1.00 to 500 ng/mL. Samples spiked with the stable isotope labeled internal standards (D6-AMG 232 and D6-AMG 232 glucuronide) were prepared by protein precipitation with acetonitrile. Extracted samples were separated by a Phenomenex Kinetex C18 analytical column (2.6 µm, 50 × 3.00 mm) with gradient elution at a flow rate of 600 µL/min, followed by electrospray ionization with negative ion multiple reaction monitoring of the parent to product ion pairs m/z 566.1→64.1 for AMG 232, m/z 574.3→64.1 for D6-AMG 232, m/z 742.5→566.0 for AMG 232 glucuronide, and m/z 750.4→574.3 for D6-AMG 232 glucuronide. Concentrations of AMG 232 and its glucuronide metabolite were calculated using a weighted 1/x2 linear regression of peak area ratios (analyte peak area/internal standard peak area) vs nominal concentrations of the calibration curve standards.
Plasma concentrations of trametinib were determined with K2EDTA anticoagulant in a validated LC-MS/MS assay using calibration curves with the range 0.100 to 250 ng/mL. Trametinib and the internal standard [13C6]-GSK1120212 were liquid-liquid extracted. After evaporation under nitrogen, the residue was reconstituted and analyzed using LC-MS/MS. Trametinib concentrations were calculated using a weighted 1/x2 linear regression calibration model.
Plasma time–concentration profiles of AMG 232 were evaluated on days 1 (0-24 h) and 7 (0-72 h). Noncompartmental analysis was performed using WinNonlin Professional software, version 6.4. Pharmacokinetic parameters were estimated, including time to maximum concentration (tmax), maximum observed plasma concentration (Cmax), area under the concentration-vs-time curve at 24 hours (AUC24h), and clearance (CL/F). Summary statistics of pharmacokinetic parameters were provided.
Biomarker analysis
Circulating MIC-1.
In both treatment arms, serum samples for the assessment of circulating MIC-1 (growth differentiation factor 15) were collected in cycles 1 and 2 on the pharmacokinetic sample schedule and end of study (≥4 weeks [or up to 7 days after] the last dose). Serum MIC-1 concentrations were measured using a commercially available ELISA kit (human growth differentiation factor 15 Quantikine, R&D Systems), per the manufacturer’s instructions.
Bone marrow expression of p53 target genes and TP53 mutational status.
In both treatment arms, bone marrow aspirates were collected at screening, 24 hours postdose on day 8 of cycle 1, postdose on day 14 of cycle 2, every 6 cycles thereafter, and at the end of treatment. For p53 target gene assessment, bone marrow aspirates collected in PAXgene Bone Marrow RNA tubes (PreAnalytiX GmbH) were stored at −20°C. RNA was extracted using the PAXgene Bone Marrow RNA Kit. RNA samples were labeled using the Low RNA Input Linear Amplification PLUS kit (Two-Color kit, Agilent Technologies). The resulting fluorescent complementary RNA was hybridized to SurePoint G3 Gene Expression Microarray 4x180K (Agilent Technologies) per the manufacturer’s instructions. Gene expression results were log2-transformed and quantile-normalized before analysis of P21 (cyclin-dependent kinase inhibitor protein), BAX (BCL2-associated X), PUMA, MDM2, and TP53. For mutational status experiments, genomic DNA from bone marrow mononuclear cells was analyzed by next-generation sequencing using the MyAML panel (Invivoscribe Technologies). TP53 mutational status was considered positive if a sample contained a somatic variant with high or moderate impact (eg, stop-gain, frameshift, or missense) on the gene product; the common TP53 SNP Pro72Arg was considered a common germline mutation and not a somatic variant. Variant impact was assessed with the SnpEff tool.26
Clinical response
Statistical analysis
Primary end points were the patient incidence of DLTs, AEs, or clinically significant or grade ≥3 changes in safety assessments and AMG 232 and trametinib pharmacokinetic parameters. Secondary/exploratory end points included best clinical response, change in serum MIC-1 level, and bone marrow expression of the p53 target genes P21, BAX, PUMA, and MDM2. Data were summarized using descriptive statistics.
Results
Patients
Thirty-six patients (arm 1, n = 26; arm 2, n = 10) with relapsed/refractory AML were enrolled between 1 April 2014 and 19 April 2017. Patient demographics and baseline characteristics are summarized in Table 1. Most patients (64%) were male, and nearly all patients (97%) were heavily pretreated and received multiple prior lines of therapy, with 42% receiving ≥3 prior lines. Four (11%) patients had received prior stem cell transplants. TP53 mutational status was known for 16 of 36 (44%) patients at enrollment who had evaluable bone marrow profiled by next-generation sequencing. Of these, 13 (36%) had no TP53 mutations, and 3 (8%) had TP53 mutations. Of 23 (64%) patients evaluable for FLT3 mutations, 3 (13%) had detectable FLT3 mutations, including 1 patient with FLT3 ITD, 1 patient with FLT3 TKD mutation, and 1 patient with FLT3 ITD and TKD mutation.
All 36 patients received ≥1 dose of AMG 232 in the dose escalation. The reasons for discontinuing AMG 232 were disease progression (n = 23), AEs (n = 4), patient request (n = 4), need for alternative therapy (n = 1), investigator decision (n = 1), patient ineligibility (n = 1), hospice care (n = 1), and withdrawn consent (n = 1). All 10 patients in arm 2 received trametinib; the reasons for discontinuation were disease progression (n = 6), patient request (n = 2), AE (n = 1), and hospice care (n = 1).
Dose escalation
No DLTs occurred in arm 1, and the MTD was not reached. The doses of AMG 232 evaluated in arm 1 were 60 mg (n = 4) 90 mg (n = 4), 180 mg (n = 5), 240 mg (n = 3), and the intermediate dose of 360 mg (n = 10). The intermediate AMG 232 dose of 360 mg (the maximum tested dose) was enrolled due to occurrence of treatment-related gastrointestinal toxicity at lower doses of AMG 232 monotherapy (antiemetic prophylaxis was not allowed per the study protocol). The doses of 480 and 960 mg were not evaluated because of toxicity. At the dose of 360 mg (n = 10), 8 patients had treatment-related gastrointestinal toxicity: diarrhea (n = 7), nausea (n = 5), vomiting (n = 2), dyspepsia (n = 1), rectal hemorrhage (n = 1), and retching (n = 1). In arm 2 (AMG 232 60 mg once daily + trametinib 2 mg once daily), 1 patient had a DLT of serious, treatment-related grade 3 fatigue on study day 4 that resolved without treatment interruption. In arm 2 (n = 10), 8 patients had treatment-related gastrointestinal toxicity: nausea (n = 8), vomiting (n = 6), diarrhea (n = 5), abdominal pain (n = 2), dyspepsia (n = 1), constipation (n = 1), and melena (n = 1). AMG 232 60 mg once daily combined with trametinib 2 mg once daily was the highest combination of doses tested in arm 2. Further dose escalation was halted due to the incidence and severity of gastrointestinal AEs at higher doses in arm 1, and the dose expansion was not enrolled.
Safety and tolerability
Thirty-five patients (97%) experienced treatment-emergent AEs (Table 2). The most common (occurring in ≥40% of patients) treatment-emergent AEs were diarrhea (83%), nausea (67%), febrile neutropenia (53%), decreased appetite (44%), fatigue (44%), and vomiting (42%). Thirty-one (86%) patients had AEs that were considered by the investigators to be attributable to treatment with AMG 232. The most common (occurring in ≥10% of patients) treatment-related AEs were nausea (58%), diarrhea (56%), vomiting (33%), decreased appetite (25%), anemia (22%), leukopenia (17%), thrombocytopenia (17%), fatigue (14%), and abdominal pain (11%). The majority (61%) of treatment-related AEs were of grade 1 or 2 in severity. Grade 3 and 4 treatment-related AEs were reported in 16 (44%) and 10 patients (28%), respectively. Grade 3 and 4 AEs of interest included leukopenia (grade 4, n = 6), thrombocytopenia (grade 3, n = 1; grade 4, n = 5), febrile neutropenia (grade 3, n = 2; grade 4, n = 1), neutropenia (grade 3, n = 1; grade 4, n = 2), platelet count decreased (grade 4, n = 2), diarrhea (grade 3, n = 2), vomiting (grade 3, n = 2), and nausea (grade 3, n = 1).
All 10 patients in arm 2 (100%) had AEs that were considered by the investigators to be attributable to treatment with trametinib. The most common (occurring in ≥20% of patients) trametinib-related AEs were nausea (70%), vomiting (70%), diarrhea (50%), anemia (30%), decreased appetite (30%), thrombocytopenia (30%), abdominal pain (20%), dysgeusia (20%), dyspnea (20%), fatigue (20%), and leukopenia (20%).
Overall, 28 patients (78%) had serious treatment-emergent AEs during the study. Among these, 7 (19%) had serious AEs that were considered related to AMG 232, including 3 with febrile neutropenia (grade 3, n = 2; grade 4, n = 1), 2 with nausea (grade 2, n = 1; grade 3, n = 1), 1 with grade 3 fatigue, 1 with grade 4 leukopenia, 1 with grade 4 neutropenia, 1 with grade 4 platelet count decreased, 1 with grade 3 pulmonary alveolar hemorrhage, and 1 with grade 3 rectal hemorrhage. Three patients had AEs resulting in discontinuation of AMG 232: grade 3 hypokalemia, grade 2 mouth ulceration and grade 2 aspartate aminotransferase increase, and pneumonia.
Seven patients had fatal AEs during the study, none of which were considered by the investigators to be related to study treatment. All occurred in patients in arm 1. Three patients in the 360-mg cohort of arm 1 died as a result of progression of relapsed AML. A patient in the 60-mg cohort of arm 1 had fatal cardiac arrest. A patient in the 360-mg cohort had fatal pneumonia. A patient in the 90-mg cohort of arm 1 had fatal respiratory failure. A patient (arm 1, 360 mg) worsening AML and low platelet count (6,000/μL) had a presumed fatal cerebral hemorrhage on study day 28 following serious rectal hemorrhage on study day 6.
Pharmacokinetics of AMG 232 and trametinib
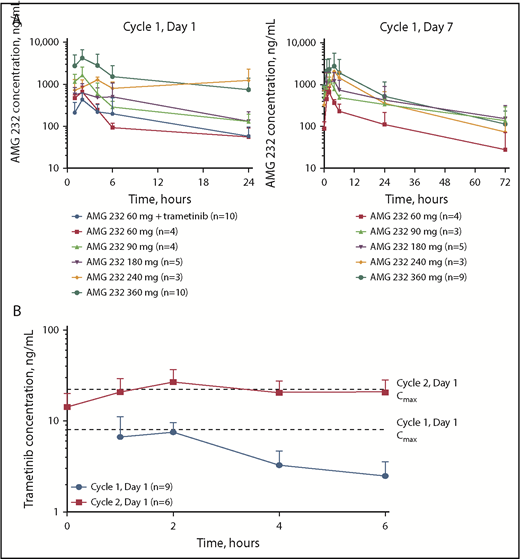
AMG 232 plasma concentration data were collected from 36 patients. Pharmacokinetic parameter estimates of AMG 232 are provided in Table 3, and the pharmacokinetic profile of AMG 232 is shown in Figure 1A. AMG 232 was absorbed rapidly, with a median tmax ranging from 2.0 to 4.0 hours across dose cohorts. The systemic AMG 232 plasma exposure, as assessed by Cmax, and AUC24h generally increased with increasing dose and at a dose of 60 mg appeared unaffected by coadministration with trametinib. The mean accumulation ratio of AUC24h ranged from 1.11 to 2.38. The mean CL/F values ranged from 7.27 L/h to 22.5 L/h and did not appear to be dependent on AMG 232 dose.

Pharmacokinetics of AMG 232 and trametinib. Mean (±standard deviation) plasma concentration vs time profiles after oral once-daily administration of AMG 232 (A) and trametinib 2 mg (B).
Pharmacokinetics of AMG 232 and trametinib. Mean (±standard deviation) plasma concentration vs time profiles after oral once-daily administration of AMG 232 (A) and trametinib 2 mg (B).
Trametinib plasma concentration data were collected from 9 and 6 patients on day 1 of cycles 1 and 2, respectively. Pharmacokinetic parameter estimates of trametinib are provided in Table 4, and the pharmacokinetic profile of trametinib over 6 hours is shown in Figure 1B. When trametinib 2 mg was coadministered with AMG 232 60 mg in arm 2, trametinib was rapidly absorbed, with median tmax values reached in <2 hours. The mean exposure ratios (day 15 vs day 1) of trametinib were 3.14-fold for Cmax and 4.33-fold for AUC6h after daily doses of 2 mg. Observed mean Cmax values (cycle 1, day 1: 8.93 ng/mL; cycle 2, day 1: 28.0 ng/mL) were consistent with those of trametinib administered as monotherapy.30,31
AMG 232 pharmacodynamic effects
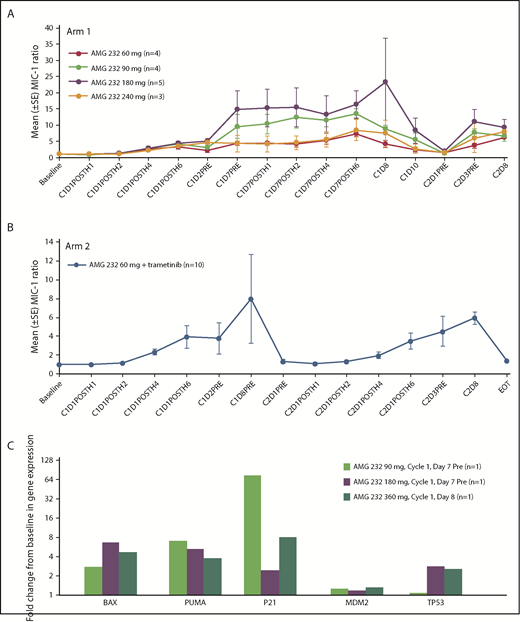
Serum MIC-1 levels were evaluable in 16 patients in arm 1 (60 mg, n = 4; 90 mg, n = 4; 180, n = 5; 240 mg, n = 3) and in 10 patients in arm 2. The serum MIC-1 fold change from baseline was variable and increased over the first 24 hours and then with repeated dosing through the end of cycle 1, followed by a decline between days 7 and 10 and a return to baseline by day 1 of cycle 1, indicating a pharmacodynamic effect by AMG 232 (Figure 2A). Similar results were observed when AMG 232 60 mg was coadministered with trametinib 2 mg (Figure 2B).

Pharmacodynamic response of MIC-1 and p53 target gene expression during treatment with AMG 232. Mean (±standard error) ratio of posttreatment vs pretreatment serum MIC-1 in arm 1 (A) and arm 2 (B). (C) Fold change from baseline in expression of BAX, PUMA, P21, MDM2, and TP53 genes in bone marrow. EOT, end of treatment.
Pharmacodynamic response of MIC-1 and p53 target gene expression during treatment with AMG 232. Mean (±standard error) ratio of posttreatment vs pretreatment serum MIC-1 in arm 1 (A) and arm 2 (B). (C) Fold change from baseline in expression of BAX, PUMA, P21, MDM2, and TP53 genes in bone marrow. EOT, end of treatment.
Expression analysis of paired pretreatment and posttreatment bone marrow was evaluable in 3 patients in arm 1 (90 mg, n = 1; 180 mg, n = 1; 360 mg, n = 1). From baseline to day 7 or 8, there was evidence of increased expression of p53 target genes, including BAX, PUMA, P21, and MDM2, as well as TP53 (Figure 2C). These data suggest that inhibition of MDM2 by AMG 232 promotes the transcriptional activation of the p53 pathway in leukemic bone marrow.
Response
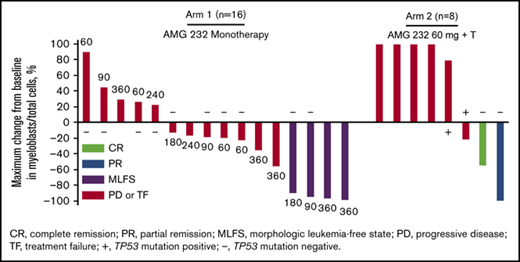
Thirty patients (83%) were evaluable for tumor response. Six patients had no bone marrow evaluation after baseline to assess response. Based on revised IWG criteria,27 one patient (3%) in arm 2 (AMG 232 + trametinib) achieved complete remission (CR), 4 patients (11%) in arm 1 (AMG 232 monotherapy) achieved morphologic leukemia-free state (MLFS; 90 mg, n = 1; 180 mg, n = 1, 360 mg, n = 2), and 1 patient (3%) from arm 2 achieved partial remission (PR) (Figure 3). Based on recommendations from an international panel on behalf of the European LeukemiaNet,28 13 patients (36%) had treatment failure as a best result and were further classified as having resistant disease (n = 11) and relapse (n = 2), whereas 11 patients (31%) had progressive disease as a best result. The patient with a best result of CR discontinued AMG 232 due to disease progression. The patient with a best result of PR discontinued AMG 232 due to needing hospice care. The 4 patients with a best result of MLFS discontinued AMG 232 due to AEs (n = 2), disease progression (n = 1), and requirement for alternative therapy (n = 1). Of 16 patients evaluated for TP53 mutational status at screening, 4 of 13 patients (31%) without TP53 mutations were responders (CR, n = 1; MLFS, n = 2; PR, n = 1), whereas none of the 3 patients (0%) with TP53 mutations were responders. FLT3 abnormalities were not detected among responders. Although MyAML next-generation sequencing was performed, there was no clear pattern associated with response to treatment due to the small number of available samples and heterogeneity of cytogenetic profiles (not shown).

Activity of AMG 232 with or without trametinib. (A) Maximum change from baseline in bone marrow blast percentage and best overall response in 24 evaluable patients in arm 1 (AMG 232 monotherapy) and arm 2 (AMG 232 + trametinib). Unevaluable patients either had no baseline measurement for bone marrow blast count or only 1 measurement in total. (B) Median treatment duration of responders (blue bars; n = 6) and nonresponders (red bars; n = 24). In both panels, dose cohorts are shown next to each bar, and TP53 mutation status, when evaluated in patients with unknown status at screening, is shown as positive (+) or negative (−). PD, progressive disease; TF, treatment failure.
Activity of AMG 232 with or without trametinib. (A) Maximum change from baseline in bone marrow blast percentage and best overall response in 24 evaluable patients in arm 1 (AMG 232 monotherapy) and arm 2 (AMG 232 + trametinib). Unevaluable patients either had no baseline measurement for bone marrow blast count or only 1 measurement in total. (B) Median treatment duration of responders (blue bars; n = 6) and nonresponders (red bars; n = 24). In both panels, dose cohorts are shown next to each bar, and TP53 mutation status, when evaluated in patients with unknown status at screening, is shown as positive (+) or negative (−). PD, progressive disease; TF, treatment failure.
Reductions from baseline in bone marrow myeloblasts as a best result occurred in 14 patients (39%), including 1 with CR, 1 with PR, 4 with MLFS, and 8 with PD or TF (Figure 3A), indicating potential evidence for activity of AMG 232 even in the absence of objective response. The patient in arm 2 who achieved CR had a duration of response of 552 days; the patient discontinued trametinib after 2 cycles due to grade 2 trametinib-related retinal detachment and AMG 232 was continued. The median duration of response (CR, partial response, and morphologic leukemia-free state) was 66 days (range, 21-552). The median time on study of responders was 100.5 days (range, 48-650). The treatment duration among responders and nonresponders is shown in Figure 3B.
Discussion
AMG 232 showed acceptable tolerability as monotherapy in a first-in-human study of patients with P53WT advanced solid tumors or multiple myeloma.7 In preclinical models, AMG 232 had shown improved antitumor effects with MEK inhibitors,10 providing the rationale for evaluating AMG 232 combination therapy. In this study in patients with relapsed/refractory AML, the AEs related to treatment with AMG 232 as monotherapy or combined with trametinib were generally mild or moderate and were consistent with the disease state and known toxicities of each agent. Based on the lack of DLTs in the monotherapy arm, the MTD was not reached. Similarly, the MTD was not reached in arm 2. One patient who received AMG 232 combined with trametinib had a DLT of AMG 232–related grade 3 fatigue that resolved without treatment interruption. Dose escalation was discontinued due to gastrointestinal AEs at higher doses in arm 1, and the dose expansion phase was not enrolled. However, gastrointestinal AEs may have been inadequately managed because prophylactic medications were not allowed per the study protocol. Ongoing and future clinical studies of AMG 232 allow use of prophylactic medications, such as ondansetron, to reduce the occurrence of nausea and vomiting and loperamide at first appearance of diarrhea.
Consistent with observations among patients who received AMG 232 monotherapy in the first-in-human study,7 the most frequent treatment-related AEs in both treatment arms were gastrointestinal toxicity (nausea, diarrhea, and vomiting), decreased appetite, anemia, leukopenia, thrombocytopenia, and fatigue. Most gastrointestinal AEs were grade 1 or 2 in severity. Serious AEs reported during the study included myelosuppression (febrile neutropenia and leukopenia) and nausea. Gastrointestinal toxicity and myelosuppression have been identified as class effects of MDM2 inhibitors.15-18,32-35 Ongoing and future studies of AMG 232 allow use of prophylactic medications for gastrointestinal toxicity. Gastrointestinal toxicity, fatigue, decreased appetite, and thrombocytopenia have also been reported during treatment with trametinib.31,36,37
AMG 232 plasma exposure increased with increasing dose. Pharmacokinetic parameters were generally similar between patients who received AMG 232 60 mg as monotherapy and those who received AMG 232 60 mg combined with trametinib. Trametinib pharmacokinetic parameters (Cmax and tmax) when coadministered with AMG 232 were consistent with published values.30,31
Activation of the p53 pathway results in the production of MIC-1,19,20 a transforming growth factor-β superfamily growth inhibitor that has been associated with poor outcomes for some cancers.38-40 In this study, the serum MIC-1 fold change from baseline was variable and increased over the first 24 hours and then declined between days 7 and 10 and returned to baseline by day 1 of cycle 1. Similar changes in MIC-1 were observed with AMG 232 administered in combination with trametinib. Increased circulating MIC-1 has been reported as a pharmacodynamic effect of treatment with other MDM2 inhibitors in patients with relapsed/refractory AML and in patients with other solid tumors.15-18,41 Although MIC-1 has been shown to contribute to the chemoprotection of AML cells,42 whether MIC-1 contributed to AML cell survival following treatment with AMG 232 in this study is unclear. Loss of p53 function in AML is often the result of overexpression of negative regulators of p53, such as MDM2.43,44 Thus, we also assessed the expression of p53 target genes using a microarray. Expression of BAX, PUMA, P21, and MDM2 increased in leukemic bone marrow following treatment with AMG 232, as has been demonstrated with other MDM2 inhibitors.15-17 These data demonstrate that inhibition of the MDM2-p53 interaction by AMG 232 leads to the upregulation of p53 transcriptional targets, consistent with the proposed mechanism of action.
Of 30 patients evaluable for response by revised IWG criteria, 1 patient (3%) who received AMG 232 combined with trametinib achieved a best response of CR, 4 (11%) achieved morphologic leukemia-free state, and 1 (3%) achieved PR. Notably, 4 of 13 patients (31%) without TP53 mutations were responders, whereas none of the 3 patients (0%) with TP53 mutations were responders, consistent with other studies of MDM2 inhibitors in AML, in which most patients evaluable for hematologic response and TP53 mutation status were P53 wild-type.15,33,35
In conclusion, in this population of patients with relapsed/refractory AML, AEs related to treatment with AMG 232 as monotherapy or combined with trametinib were generally mild or moderate in severity and were consistent with the known toxicities of each agent, as well as the disease state. No DLTs occurred in the AMG 232 monotherapy arm, and the MTD was not determined. AMG 232 dose escalation to doses >360 mg was not performed due to the occurrence of gastrointestinal AEs at higher doses. In arm 2, the MTD of AMG 232 was 60 mg once daily when combined with trametinib 2 mg once daily. AMG 232 pharmacokinetics were linear with increasing dose. Exposure to AMG 232 was not affected by coadministration with trametinib. AMG 232 treatment, either as monotherapy or in combination with trametinib, resulted in on-target biological effects and was associated with clinical activity, with 1 patient with CR, 1 with PR, and 4 with MLFS. Of patients tested for TP53 mutations, 31% without TP53 mutations were responders and none with mutations were responders. Future clinical studies of AMG 232 in AML and in other hematologic indications are under consideration. AMG 232 is currently under clinical development as KRT-232 in myelofibrosis (NCT03662126), polycythemia vera (NCT03669965), and Merkel cell carcinoma (NCT03787602).
Presented as a poster at the American Society of Clinical Oncology Annual Meeting, Chicago, IL, 2-6 June 2017.
Acknowledgments
The authors thank Ben Scott (Scott Medical Communications, LLC) for medical writing assistance funded by Amgen Inc.
This study was funded by Amgen Inc.
Authorship
Contribution: H.P.E. designed the study, collected data and enrolled patients, interpreted data, and wrote the manuscript; P.S.B., P.J.S., M.R.G., and E.S.W. collected data and enrolled patients, interpreted data, and wrote the manuscript; D.L.F., M.Z., E.R., and A.A.A. interpreted the data, performed statistical analysis, and wrote the manuscript; and H.A.H. designed the study, interpreted the data, and wrote the manuscript.
Conflict-of-interest disclosure: H.P.E. is a consultant/advisor for Amgen Inc., Celator/Jazz, Celgene, Daiichi Sankyo, Glycomimetics, ImmunoGen, Incyte, MacroGenics, Millenium/Takeda, Novartis, Ono, Pfizer, Seattle Genetics, and Sunesis; a member of the speakers’ bureau for Agios, Celgene, Incyte, and Novartis; receives research funding from Agios, Amgen Inc., Astellas, Celator, Daiichi Sankyo, ImmunoGen, Janssen, Juno, Millenium/Takeda, and Seattle Genetics; and is on the data safety monitoring board for Glycomimetics and the steering committee for Celgene. P.S.B. is a consultant/advisor for Pfizer and Caremark and receives research funding from Amgen Inc., Glycomimetics, AbbVie, JW Pharmaceuticals, Bristol-Myers Squibb, Novartis, and Trovagene. P.J.S. has stock ownership in JSK Therapeutics and Lone Star Therapeutics; is a consultant/advisor for Tolero, Pfizer, and Baxalta; and receives research funding from Cantex and Amgen Inc., patents/royalties from JSK Therapeutics, and research support from Pfizer. M.R.G. has stock ownership in Medtronic; is a consultant/advisor for Incyte, Alexion, Amgen Inc., Pfizer, Merck, Agios, Cardinal Health, Ariad, Celgene, and AbbVie; and receives research funding from Incyte, Genentech/Roche, Amgen Inc., Janssen, and Forma Therapeutics. D.L.F., M.Z., E.R., H.A.H., and A.A.A. are employed by and have stock ownership in Amgen Inc. E.S.W. is a consultant/advisor for AbbVie, Arog, Immunogen, Pfizer, and Amgen Inc.; a member of speakers’ bureau for Jazz Pharmaceuticals and Novartis; and receives research funding from Immunogen.
Correspondence: Harry P. Erba, Division of Hematologic Malignancies and Cellular Therapy, Department of Medicine, Duke University, 2400 Pratt St, Suite 5000, Durham, NC 27705; e-mail harry.erba@duke.edu.