Key Points
In patients with an MRD− treatment response, we found a higher abundance of the butyrate producer E hallii.
Deep response to MM therapy is a biomarker for progression-free survival, suggesting association of microbial signature and patient outcome.

Abstract
Patients with multiple myeloma (MM) who achieve minimal residual disease (MRD) negativity after upfront treatment have superior outcomes compared with those who remain MRD+. Recently, associations have been shown between specific commensal microbes and development of plasma cell disorders. Here, we report the association between intestinal microbiota composition and treatment outcome in MM. Microbiota composition of fecal samples collected from 34 MM patients after induction therapy and at the time of flow cytometry–based bone marrow MRD testing was determined by 16S ribosomal RNA sequencing. We observed a higher relative abundance of Eubacterium hallii in the 16 MRD− patients relative to the 18 MRD+ patients. No association was observed between microbial relative abundance and autologous stem cell transplantation history or MM paraprotein isotype. No differences in microbiota α diversity were observed between MRD− and MRD+ patients. The potential association of microbiota composition with treatment response in MM patients is an important parameter for additional correlative and clinical investigation.
Introduction
Multiple myeloma (MM) is a malignant plasma cell disorder that is highly responsive to first-line therapy. However, relapses with drug-resistant disease clones cause disease-related mortality. Newly diagnosed MM patients who achieve deep responses, characterized by minimal residual disease (MRD) negativity after upfront treatment, have superior outcomes compared with those who remain MRD+. The proportion of patients reaching MRD negativity is increasing with newer combination therapies.1 As such, MRD has become a clinically relevant end point in the development of modern therapies for MM.2
The gut microbiome has been implicated in numerous aspects of human health, including in the development of certain cancers, treatment response, and treatment-related toxicity.3 A commensal member of both the human and murine microbiota, Prevotella heparinolytica has promoted MM progression via interleukin-17 (IL-17)– and eosinophil-mediated inflammation in a mouse model of myelomagenesis.4 In humans, microbes of the genus Eubacterium have been associated with reduced risk of MM relapse after allogeneic hematopoietic cell transplantation.5 Studies evaluating the relationships between commensal microbiota, treatment, and/or toxicity in MM patients have yet to be performed.
Herein, we evaluate the relationship between intestinal microbiota and treatment outcome in MM by metagenomic analysis of prospectively collected fecal samples from patients who have completed induction therapy.
Methods
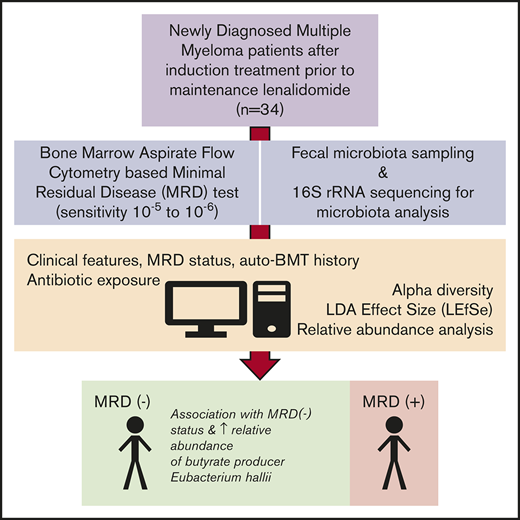
MM patients who completed first-line therapy and were candidates for lenalidomide maintenance were enrolled prospectively in accordance with the Declaration of Helsinki under a protocol approved by our institutional review board. Disease responses were assessed according to International Myeloma Working Group guidelines and criteria.6 Patients were evaluated for MRD status after completion of induction and before start of lenalidomide maintenance therapy using a validated bone marrow–based flow cytometric assay with a sensitivity of at least 10−5.7,8 Exposure to antibiotics within 60 days of stool collection was captured by patient survey and chart review. Stool samples were collected without additives, refrigerated in an insulated container, processed within 24 hours, and stored frozen at −80°C until sequencing. DNA extracted from fecal samples was subjected to amplification of the V4/V5 variable regions of the 16S ribosomal RNA genes using barcoded primers targeted to conserved sequences.9,10 Purified polymerase chain reaction products were sequenced on the Illumina MiSeq platform, and 16S paired-end reads were merged and demultiplexed. The UPARSE pipeline was used to perform quality and error filtering using maximum expected error (Emax = 1), and reads with >97% identity were grouped into operational taxonomic units (OTUs).11 A phylogenetic tree was constructed from a sequence alignment of all OTUs. All analyses were performed using R (version 3.5.0; R Development Core Team, Vienna, Austria) and the phyloseq software package.12 We used the linear discriminant analysis effect size (LEfSe) method to analyze relationships between microbiota and MRD status using autologous stem cell transplantation as a subclass parameter.13 LEfSe output was filtered for species present in 10 patients with average relative abundance >0.01 and subjected to Benjamini-Hochberg false discovery rate (FDR) correction. Taxa with FDR <0.10 were considered sufficiently promising. Comparison of α diversity metrics was assessed using the Wilcoxon rank sum test. Unsupervised sample clustering was performed via principal coordinates analysis, with dissimilarity assessed using a weighted UniFrac distance metric.14 Raw data are available upon request.
Results and discussion
We enrolled 42 MM patients between January 2017 and April 2018. Eight patients received salvage therapy for relapsed disease before enrollment on study. The remaining 34 patients met entry criteria for the post–induction therapy analysis. Baseline characteristics of the postinduction cohort are listed in Table 1. The median age of the 34 patients was 62.5 years (range, 38-84 years), and 13 (38.2%) were women. Sixteen (47.1%) of 34 patients were MRD− at baseline. First-line therapy included carfilzomib, lenalidomide, and dexamethasone in 24 patients (70.6%) and autologous stem cell transplantation in 14 patients (41.2%).
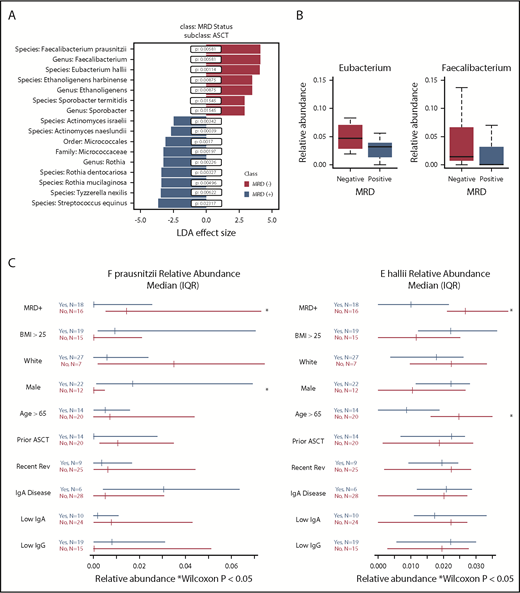
In 34 sequenced stool samples, 479 OTUs with >100 sequences were identified and mapped to 8 phyla, 149 genera, and 301 species. There was no significant difference in α diversity, as measured by theiInverse Simpson index, between MRD− (median, 12.24; interquartile range [IQR], 8.76-13.98) and MRD+ patients (median, 12.44; IQR, 8.36-16.23; P = .6; supplemental Figure 1). Samples differed by MRD status based on principal component analysis (supplemental Figure 2). Using LefSe analysis, we found a positive association between MRD− status, with 3 taxa identified at the genus level and 4 bacterial species. MRD− status was negatively associated with 1 taxon identified at the order and family level, 1 taxon identified at the genus level, and 6 bacterial species (Figure 1A). Filtering on bacteria present in at least 10 patients resolved the output to 2 butyrate producers, Eubacterium hallii (P = .00114; FDR, 0.0913) and Faecalibacterium prausnitzii (P = .00581; FDR, 0.2322). Thus, E hallii remained significant after FDR correction.
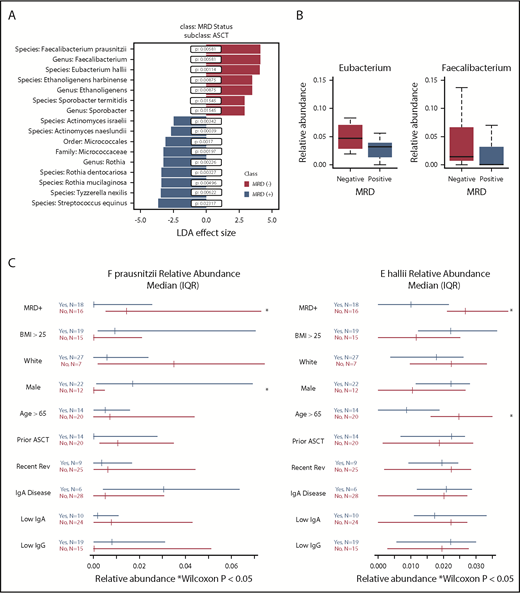
Fecal microbiota analysis according to MRD status. (A) Linear discriminant analysis (LDA) effect size analysis of microbiota differentially associated with MRD status with subclass of autologous stem cell transplantation (ASCT). (B) Relative abundance of genera Eubacterium and Faecalibacterium by MRD status. (C) Forest plots showing effect of covariates on relative abundance of E hallii and F prausnitzii.
Fecal microbiota analysis according to MRD status. (A) Linear discriminant analysis (LDA) effect size analysis of microbiota differentially associated with MRD status with subclass of autologous stem cell transplantation (ASCT). (B) Relative abundance of genera Eubacterium and Faecalibacterium by MRD status. (C) Forest plots showing effect of covariates on relative abundance of E hallii and F prausnitzii.
Because F prausnitzii and E hallii were identified in LeFse analysis and present in ≥10 patients, we sought to further evaluate these 2 taxonomic groups. We performed a differential abundance analysis of these selected taxa in MRD+ and MRD− patients at the genus and species levels. Relative abundances of genus Eubacterium and genus Faecalibacterium were higher in MRD− compared with MRD+ patients (Eubacterium: MRD−: median, 4.71%; IQR, 2.88%-6.98% vs MRD+: median, 3.17%; IQR, 1.44%-3.84%; Faecalibacterium: MRD−: median, 1.43%; IQR, 0.77%-6.43% vs MRD+: median, 0.003%; IQR, 0%-2.55%; Figure 1B). Both E hallii and F prausnitzii were enriched in MRD− compared with MRD+ patients (E hallii: MRD− : median, 3.16%; IQR, 2.11%-5% vs MRD+: median, 1.01%; IQR, 0%-2.16%; F prausnitzii: MRD−: median, 1.43%; IQR, 0.77%-6.43% vs MRD+: median, 0.003%; IQR, 0%-2.55%). Covariates associated with differential abundance of E hallii and F prausnitzii revealed an association with age <65 years and sex, respectively, for the 2 species (Figure 1C). Sex hormone levels were not assessed on study. In a post hoc analysis, relapsed vs postinduction status was not associated with differential abundance of these 2 microbes (data not shown). We did not observe an association with treatment type, immunoglobulin isotype, or serum immunoglobulin concentrations.
F prausnitzii is a common butyrate-producing commensal microbe often associated positively with human gut health.14 Butyrate and other short-chain fatty acids, such as propionate, also produced by E hallii,15 are biologically active metabolites formed during microbial fermentation of dietary or host-derived carbohydrates, which supply the host with energy and also modulate immunity via exertion of anti-inflammatory functions.15-18 Butyrate is known to inhibit in vitro production of IL-17A via downregulation of Th17 cells in human peripheral blood mononuclear cells.19 Of note, IL-17–producing Th17 cells induced by microbiota have been associated with disease pathogenesis in a murine MM model,4 and after bone marrow transplantation, promotion of MM immune escape is mediated by donor-derived IL-17A.20
In conclusion, we observed a higher relative abundance of the butyrate producer E hallii in MRD− MM patients compared with MRD+ MM patients. We additionally identified F prausnitzii as another microbe potentially associated with MRD− status after initial therapy for MM. Our results are novel and support the concept that intestinal microbiota composition is associated with deep treatment response. Future studies are needed to confirm our findings in larger cohorts and enable additional characterization of the relationships between disease characteristics (eg, isotype or cytogenetics) vs overall health status in microbial composition in the gut as it relates to treatment response. More broadly, preclinical studies are needed to better understand the molecular relationship between microbiome signature and clinical outcome in myeloma as well as other hematologic malignancies.
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by National Institutes of Health awards U01-AI124275, R01-AI095706, and R01-AI42135 (E.G.P) from the National Institute of Allergy and Infectious Diseases and awards P01-CA023766 (M.R.M.v.d.B. and E.G.P.), R01-CA228308 (M.R.M.v.d.B.), R01-CA228358 (M.R.M.v.d.B.), and L30-CA220724 (M.J.P.) from the National Cancer Institute; Memorial Sloan Kettering (MSK) Cancer Center (MSKCC) Support Grant/Core Grant award P30-CA008748; National Institute on Aging award Project 2 of P01-AG052359 (M.R.M.v.d.B.); National Center for Advancing Translational Sciences award UL1-TR00457 (M.J.P.); Cycle for Survival to the MSK Center for Microbes, Inflammation and Cancer; the MSK Sawiris Foundation; the Parker Institute for Cancer Immunotherapy at MSKCC (J.U.P., M.R.M.v.d.B., and A.M.L.); The Lymphoma Foundation; The Susan and Peter Solomon Divisional Genomics Program; the Mortimer J. Lacher Fellowship Fund of the Lymphoma Foundation of MSKCC (M.J.P.); and Seres Therapeutics (J.U.P. and M.R.M.v.d.B).
Authorship
Contribution: M.J.P. designed the study and conducted data collection, data analysis, data interpretation, and manuscript preparation; E.R.L. assisted in data analysis and interpretation and drafted the figures; E.F. and L.L. conducted total 16S quantitative polymerase chain reaction and conducted microbiota sequencing; S.M.D. assisted in data analysis and statistical interpretation and methods; A. Chansakul, D.M., M.S., E.F., L.L., E.T., J.B.S., A.E.S., and A. Clurman assisted in procurement, storage, processing, and collection of samples; M.J.P., S.M.D., E.R.L., A.L.C.G., Y.T., E.G.P., J.U.P., M.R.M.v.d.B., and O.L. assisted in data analysis, data interpretation, and manuscript preparation; A.M.L. designed the study, supervised data collection and data interpretation, and conducted manuscript preparation; and all authors critically reviewed and approved the manuscript.
Conflict-of-interest disclosure: J.U.P. reports intellectual property (IP) licensing with and research funding from Seres Therapeutics. A. Clurman, J.B.S., A.E.S., and A.L.C.G. report research funding from Seres Therapeutics. M.R.M.v.d.B. reports research support from Seres Therapeutics, honoraria from or consultancy or advisory board participation for Seres Therapeutics, Flagship Ventures, Novartis, Evelo, Jazz Pharmaceuticals, Therakos, Amgen, Merck, Acute Leukemia Forum, and DKMS Medical Council, and IP licensing with Seres Therapeutics and Juno Therapeutics. O.L. reports consultancy for Janssen, Merck, Pfizer, Takeda, Karyopharm, Amgen, Celgene, Binding Site, Adaptive, Cellectis, Glenmark, and Juno Therapeutics, honoraria from Janssen, Pfizer, Karyopharm, Amgen, Celgene, Binding Site, and Adaptive, and research funding from Janssen, Takeda, Amgen, Celgene, Glenmark, and Seattle Genetics. A.M.L. reports consultancy for Genmab, Bristol-Myers Squibb, and Takeda, honoraria from Genmab, Bristol-Myers Squibb, and Takeda, research funding from Genentech, Bristol-Myers Squibb, and Janssen, and royalties from Serametrix. The remaining authors declare no competing financial interests.
Correspondence: Alexander M. Lesokhin, Myeloma Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, 1233 York Ave, New York, NY 10065; e-mail: lesokhia@mskcc.org.