

Key Points
We report the first known family with a constitutional translocation disrupting ETV6 predisposing to ALL.
Germline monoallelic expression of ETV6 contributes to leukemia predisposition without thrombocytopenia.

Abstract
Pathogenic germline variants in ETV6 have been associated with familial predisposition to thrombocytopenia and hematological malignancies, predominantly childhood B-cell precursor acute lymphoblastic leukemia (BCP-ALL). In addition, overrepresentation of a high hyperdiploid subtype and older age at diagnosis have been reported among sporadic BCP-ALL cases with germline variants in ETV6. We studied a family with 2 second-degree relatives who developed childhood high hyperdiploid BCP-ALL at ages 8 and 12 years, respectively. A constitutional balanced reciprocal translocation t(12;14)(p13.2;q23.1) was discovered in both patients by routine karyotyping at diagnosis and, subsequently, in 7 healthy family members who had not experienced hematological malignancies. No carriers had thrombocytopenia. Whole-genome sequencing confirmed the translocation, resulting in 2 actively transcribed but nonfunctional fusion genes, causing heterozygous loss and consequently monoallelic expression of ETV6. Whole-genome sequencing analysis of the affected female subjects’ leukemia excluded additional somatic aberrations in ETV6 and RTN1 as well as shared somatic variants in other genes. Expression studies, performed to confirm decreased expression of ETV6, were not conclusive. We suggest that germline aberrations resulting in monoallelic expression of ETV6 contribute to leukemia susceptibility, whereas more severe functional deficiency of ETV6 is required for developing THC5. To our knowledge, this report is the first of a constitutional translocation disrupting ETV6 causing predisposition to childhood ALL.
Introduction
The etiology of childhood acute lymphoblastic leukemia (ALL) is unknown in the majority of cases.1 However, compelling evidence for the contribution of genetic predisposition to childhood ALL in congenital intellectual disability and/or malformation syndromes with increased risk of cancer2,,,,,,,-10 and de novo or inherited cancer predisposition syndromes11,,,,,,,,,,,,-24 has emerged. Recently, several families carrying rare germline variants in ETS variant 6 (ETV6) with familial thrombocytopenia and predisposition to hematological malignancies have been reported (THC5; MIM#616216).25,,,,,,-32 Most assayed variants have been found to act through dominant negative repression of wild-type ETV6.29,32,33 In addition, Moriyama et al28 reported 35 children with B-cell precursor ALL (BCP-ALL), with no known family history of hematological malignancies, to have germline variants in ETV6 potentially responsible for leukemia predisposition. The majority of these cases had a high hyperdiploid (HeH) karyotype (80%), older age at diagnosis (13.3 vs 6.8 years without risk variants), and leukocyte counts <50 × 109/L at diagnosis.28,33 However, the additional somatic aberrations required for leukemic development in these cases are unknown.
In the current study, we report germline and somatic variants in a family with an inherited translocation disrupting ETV6, a balanced reciprocal t(12;14)(p13.2;q23.1), segregating with predisposition to childhood BCP-ALL.
Subjects and methods
Patients
Two female subjects (the proband and her maternal aunt) from a Finnish family were treated for childhood BCP-ALL at Oulu University Hospital in Oulu, Finland. DNA was extracted from peripheral blood (remission) and bone marrow (leukemia diagnosis) of the affected individuals, and from peripheral blood (germline) of 7 additional family members who were included in the study. DNA extraction was performed immediately after sampling and stored appropriately at −20°C until the molecular analyses. Karyotyping with G-banding (Giemsa staining) was performed on 11 individuals in the family (I:2 and I3; II:1 and II5; III:2, III3, III5, and III6; IV:1, IV2, and IV3) (Figure 1) at the University Hospital of Oulu, Finland, according to standard methods. Routine interphase and metaphase fluorescence in situ hybridization (FISH) analyses were performed on the diagnostic bone marrow sample of one individual (IV:2) using locus-specific probes for MLL rearrangements, t(1;19), t(9;22), and t(12;21) translocations, respectively.

Pedigree of the family with the t(12;14)(p13.2;q23.1) predisposing to childhood ALL.
Pedigree of the family with the t(12;14)(p13.2;q23.1) predisposing to childhood ALL.
The study was approved by the Regional Ethics Committee of Northern Ostrobothnia Hospital District, Oulu, Finland (EETTMK: 45/2015), and the Regional Ethics Committee of Karolinska Institutet/Karolinska University Hospital, Stockholm, Sweden (Dnr 2015/293-31/4). The study was performed in accordance with the 1975 Declaration of Helsinki, as revised in 2000. Written informed consent was obtained from the included individuals or their legal guardians.
Whole-genome sequencing and bioinformatics analysis
Whole-genome sequencing (WGS) was performed on genomic DNA from remission collected from the affected female subjects (III:3 and IV:2) 24 and 5 years, respectively, after leukemia diagnosis and on germline genomic DNA from one unaffected carrier (III:2). The objective was to define the exact coordinates and sequence of the translocation breakpoints and to detect other potential predisposing variants. WGS was also performed on genomic DNA from bone marrow at leukemia diagnosis in both affected female subjects (IV:2 and III:3) to characterize somatic events in the leukemic clones.
Libraries for sequencing on Illumina HiSeq X (Illumina, San Diego, CA) were prepared from the genomic DNA by using the Illumina TruSeq PCR-free kit with a mean insert size of >350 base pairs (bp). This method resulted in an average of 500 million mapped unique sequences per sample (range between 386 million and 740 million) with a mean coverage of 40× (range between 32× and 61×). An in-house pipeline developed by Science for Life Laboratory (Stockholm, Sweden) was used to map reads to the human reference genome (GRCh37/hg19) and to call variants. Data were aligned to the reference genome by using Burrows-Wheeler Aligner (version 0.7.12).34 The raw alignments were then de-duplicated, recalibrated, and cleaned by using the Genome Analysis Tool Kit (version 3.3-0-geee94ec).35 The quality control information was gathered by using Qualimap (version 2.0).36
Somatic variants were identified by using a somatic single nucleotide polymorphism (SNP) and indel caller MuTect2.37 Variant files were later manipulated by using the CatVariants and SelectVariants tools in the Genome Analysis Tool Kit. The somatic variants were functionally annotated by using the Variant Effect Predictor (version 89)38 and loaded into a database using GEMINI (version 0.20.0).39 The variants were explored in the database by using built-in tools in GEMINI and visualized in the Integrative Genomics Viewer (IGV).40
Structural variants were detected by using the FindSV pipeline (https://github.com/J35P312/FindSV), which merges calls from CNVnator V0.3.2 and TIDDIT.41,42 The Variant Call Format (VCF) file for structural variants was annotated by using the Variant Effect Predictor and filtered based on the quality flag of the VCF file. The filtered and annotated VCF file was then sorted based on a local structural variant frequency database consisting of 400 patient samples. The reads at breakpoints were visualized in IGV. The exact coordinates of the breakpoints were identified by aligning split reads to the reference genome using the BLAST-like alignment tool (BLAT) in the UCSC Genome Browser.43,44
Sanger sequencing
Sanger sequencing was performed by using ABI3730xl DNA Analyzer (Thermo Fisher Scientific, Waltham, MA) and BigDye Terminator version 3.1 (Thermo Fisher Scientific) to validate translocation breakpoints and fusion gene transcript sequences in remission/germline samples. Primers were designed by using WGS data and in silico predictions of fusion gene transcripts, respectively. Coding regions of ETV6 were also amplified and sequenced in diagnostic bone marrow and remission samples of the 2 affected female subjects (IV:2 and III:3) and an unaffected carrier (III:2) to detect somatic aberrations. Primer sequences are available upon request.
SNP array
Both BCP-ALL cases reported here were analyzed for copy number alterations (CNAs) using Infinium Omni2.5Exome BeadChip SNP arrays (Illumina). A panel of internal controls was used to normalize probe intensities in our analysis to normal diploid samples, to generate log-transformed ratios (logR ratios), which were centered around zero for diploid samples. CNAs were called by using circular binary segmentation45 and Tumor Aberration Prediction Suite,46 as implemented in R (R Foundation for Statistical Computing). Variants <20 kb or 10 consecutive probes were filtered out, and all putative somatic variants were visualized and manually reviewed in IGV.40 Karyotypes of the leukemic clones were defined from the genome-wide profile of the logR ratios.
Expression analysis
Expression of wild-type ETV6 and RTN1 messenger RNA, as well as the fusion transcripts ETV6-RTN1 and RTN1-ETV6, was studied by using quantitative real-time polymerase chain reaction (qPCR) on a Quant-Studio 7 Flex Real-Time PCR System using Power SYBRGreen (both Thermo Fisher Scientific). Wild-type transcript primers were designed spanning exon–exon boundaries to ensure wild-type specific binding. Fusion transcript primers were designed spanning the respective fusion breakpoints. Expression of human ribosomal RNA 18S was analyzed for each sample as an internal control. All samples were run in triplicate.
For germline expression of wild-type ETV6 and RTN1, total RNA from peripheral blood was analyzed in the 2 affected female subjects (IV:2 and III:3) and one unaffected carrier (III:2) alongside 6 unrelated healthy control subjects (3 sex- and age-matched to each of the affected female subjects). Expression levels were calculated and presented as folds of control [(Etarget)ΔCPtarget(control-sample)]/[(Ereference)ΔCPreference(control-sample)], normalizing against 18S and PCR efficacy. The control value was calculated as an average of all 6 analyzed normal control samples and set to one (y = 1).
For expression of wild-type ETV6 and RTN1 in leukemias, total RNA from the diagnostic bone marrow of the 2 affected female subjects (IV:2 and III:3) were analyzed together with 8 unrelated cases of sporadic BCP-ALL: 2 ETV6 deletions (delETV6), 3 t(12;21)+delETV6, and 3 wild-type for ETV6. Expression levels were calculated and presented as ΔCt (Ct[reference] – Ct[target]), normalizing against 18S.
Results
Clinical characterization
A female proband (IV:2) and her maternal aunt (III:3) were diagnosed with childhood HeH BCP-ALL at age 12 and 8 years, respectively (Figure 1). The maternal aunt (III:3) was treated according to the Nordic Society of Pediatric Haematology and Oncology (NOPHO) high-risk ALL-92 protocol47 because of myeloid cell surface markers on leukemic blast cells, and the proband (IV:2) was treated according to the NOPHO standard-risk ALL-2008 protocol.48 They are both in complete first remission 24 years and 5 years after diagnosis, respectively. Both affected female subjects developed osteonecrosis as a severe adverse effect of chemotherapy. In addition, the proband (IV:2) developed vincristine neuropathy and osteoporosis with compression fractures (Table 1). No thrombocytopenia, abnormalities of the red blood cell lineage, or hematological malignancies, other than BCP-ALL, were reported in the family. The blood cell values were within normal range in all translocation carriers for which values were available (n = 7) (supplemental Table 1).
Three individuals in the family have been diagnosed with solid tumors (Table 2). One case of mesothelioma (II:1) and 1 case of prostate cancer (II:5) were diagnosed in 2 male translocation carriers at 62 years and 71 years of age, respectively. The exposure history in the male subject with mesothelioma (II:1) is unknown. Both individuals died of the disease 2 years after diagnosis; we have been unable to acquire germline or tumor samples for the purpose of our study. A young male subject (III:14), whose father was a carrier of the t(12;14), was diagnosed with an unspecified kidney tumor at the age of 7 years and died of relapse 10 years later. Unfortunately, no genetic material or detailed clinical information was available in this case. Migraine, asthma, and atopy (allergies and atopic eczema) were additional phenotypic characteristics reported in the family. Asthma and atopic syndrome were only reported in the translocation carriers. Migraine did not segregate consistently with the t(12;14).
The maternal aunt (III:3) developed hypogammaglobulinemia after the leukemia diagnosis and chemotherapy. She had slightly subnormal levels of immunoglobulin G (IgG) subclass IgG1 and IgG2 (supplemental Table 2). Her daughter (IV:3), also a translocation carrier, had low levels of IgG at 1 year of age without need for replacement therapy. Nevertheless, the immunoglobulin levels can vary at a young age. Follow-up immunoglobulin values for the daughter (IV:3) were not available. Reduced immunoglobulin levels were not reported in any other translocation carrier. Thus, there is no clear indication of an inherited primary immunodeficiency as stated elsewhere.49
Genetic analysis of germline aberrations
A constitutional balanced reciprocal translocation t(12;14)(p13.2;q23.1) (Figure 2A) was discovered in the maternal aunt (III:3) by routine karyotyping at the time of the leukemia diagnosis. Subsequently, the proband (IV:2), known to carry the translocation, was also diagnosed with leukemia. Karyotyping of additional family members initially occurred in conjunction with the discovery of the translocation in the maternal aunt (III:3) and was later performed on younger family members (IV:1, IV:2, and IV:3). After the diagnosis of the proband, karyotyping was performed on additional family members (II:5, III:5, and III:6) eventually confirming 7 carriers (Figure 1).
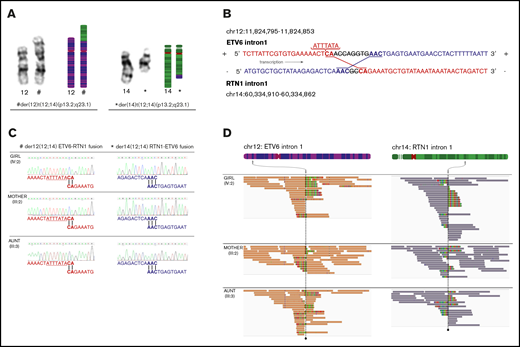
Translocation t(12;14)(p13.2;q23.1). (A) Karyotype images and models of chromosome pairs 12 and 14. The derivate chromosomes, der(12)t(12;14)(p13.2;q23.1)c and der(14)t(12;14)(p13.2;q23.1)c, created by the translocation t(12;14)(p13.2;q23.1), are indicated by # and *. (B) Model of genetic events at breakpoints and fusions. ETV6 and RTN1 are transcribed from the positive strand and negative strand, respectively. ETV6 intron 1 (positive strand) has fused to RTN1 intron 1 (negative strand) creating the chimeric fusion gene ETV6-RTN1 (indicated in red). RTN1 intron 1 (negative strand) has fused to ETV6 intron 1 (positive strand) creating the chimeric fusion gene RTN1-ETV6 (indicated in blue). An 8 bp deletion in ETV6 and a 2 bp deletion in RTN1 have occurred at the breakpoints before fusion. Microhomologies, CA and AAC, were found at the fusion points of each derivate chromosome. A 7 bp insertion was found upstream of the fusion in ETV6 intron 1. (C) Sanger traces of genomic sequence from ETV6-RTN1 and RTN1-ETV6 chimeric fusion genes, supporting identical breakpoints in affected female subjects (III:3 and IV:2) and unaffected carrier (III:2). (D) Germline and remission WGS data from affected female subjects (III:3 and IV:2) and unaffected carrier (III:2) visualized in IGV40,60 showing paired-end reads at translocation breakpoint sites on chromosomes 12 and 14. Coloring of reads (chr12 orange and chr14 purple) indicate that the 2 mates in a read-pair are mapping to different chromosomes, in this case chromosome 12 (ETV6 intron 1) and chromosome 14 (RTN1 intron 1). Multi-coloring of reads indicates mismatched nucleotides. Read-pairs mapping to 2 different chromosomes and mismatched bases on either side of the breakpoint support the presence of translocation breakpoints at either position. Gray arrows indicate the chromosomal regions where breakpoints are found. The genomic positions of breakpoints and sequences at fusion sites are identical in all 3 individuals analyzed.
Translocation t(12;14)(p13.2;q23.1). (A) Karyotype images and models of chromosome pairs 12 and 14. The derivate chromosomes, der(12)t(12;14)(p13.2;q23.1)c and der(14)t(12;14)(p13.2;q23.1)c, created by the translocation t(12;14)(p13.2;q23.1), are indicated by # and *. (B) Model of genetic events at breakpoints and fusions. ETV6 and RTN1 are transcribed from the positive strand and negative strand, respectively. ETV6 intron 1 (positive strand) has fused to RTN1 intron 1 (negative strand) creating the chimeric fusion gene ETV6-RTN1 (indicated in red). RTN1 intron 1 (negative strand) has fused to ETV6 intron 1 (positive strand) creating the chimeric fusion gene RTN1-ETV6 (indicated in blue). An 8 bp deletion in ETV6 and a 2 bp deletion in RTN1 have occurred at the breakpoints before fusion. Microhomologies, CA and AAC, were found at the fusion points of each derivate chromosome. A 7 bp insertion was found upstream of the fusion in ETV6 intron 1. (C) Sanger traces of genomic sequence from ETV6-RTN1 and RTN1-ETV6 chimeric fusion genes, supporting identical breakpoints in affected female subjects (III:3 and IV:2) and unaffected carrier (III:2). (D) Germline and remission WGS data from affected female subjects (III:3 and IV:2) and unaffected carrier (III:2) visualized in IGV40,60 showing paired-end reads at translocation breakpoint sites on chromosomes 12 and 14. Coloring of reads (chr12 orange and chr14 purple) indicate that the 2 mates in a read-pair are mapping to different chromosomes, in this case chromosome 12 (ETV6 intron 1) and chromosome 14 (RTN1 intron 1). Multi-coloring of reads indicates mismatched nucleotides. Read-pairs mapping to 2 different chromosomes and mismatched bases on either side of the breakpoint support the presence of translocation breakpoints at either position. Gray arrows indicate the chromosomal regions where breakpoints are found. The genomic positions of breakpoints and sequences at fusion sites are identical in all 3 individuals analyzed.
WGS confirmed the heterozygous germline t(12;14)(p13.2;q23.1) with identical breakpoints in intron 1 of ETV6 (chromosome 12) and RTN1 (chromosome 14), respectively, in all 3 analyzed carriers (III:2, III:3, and IV:2) (Figure 2B). Breakpoints were validated by Sanger sequencing (Figure 2C-D). The translocation resulted in 2 fusion genes, ETV6-RTN1 and RTN1-ETV6. Both transcripts are affected by a frameshift downstream of the fusion point, leading to altered codons for translation and, consequently, premature stop codons (Figure 3A). The predicted transcript sequences in and around the fusions were confirmed by using Sanger sequencing. Expression of fusion transcripts was validated by PCR amplification (Figure 3B) and qPCR (data not shown). In silico exploration of the theoretical fusion proteins showed that no functional domains were retained (Figure 3C), and no commonly occurring functional domains had emerged as a result of the frameshift.

Chimeric fusion gene transcripts ETV6-RTN1 and RTN1-ETV6. (A) In silico prediction of amino acid sequences encoded by fusion transcripts. ETV6-RTN1 fusion transcript, originating from exon 1 of ETV6 and part of exon 2 of RTN1, houses a frameshift downstream of the fusion point, resulting in a premature stop codon. The transcript is predicted to encode 86 amino acids. RTN1-ETV6 fusion transcript, originating from exon 1 of RTN1 and exons 2, 3, and part of 4 of ETV6, also houses a frameshift downstream of the fusion point resulting in a premature stop codon. This transcript is predicted to encode 178 amino acids. Electropherograms of novel exon–exon junctions, resulting from the translocation, support the in silico–predicted sequences of fusion transcripts. (B) Gel images (2% agarose gel) supporting the expression of both fusion transcripts in remission and leukemia from the 2 affected female subjects (III:3 and IV:2) and germline of an unaffected carrier (III:2). Positive control ribosomal RNA 18S5N is shown in the bottom gel image. 100bp Plus DNA Ladder from GeneRuler (Thermo Fisher Scientific). (C) Two-dimensional models of ETV6 and RTN1 protein with approximate locations of breakpoints indicated by red bars. Functional domains are not retained in theoretical fusion proteins. Breakpoints are located upstream of functional domains, and all sequence downstream of breakpoints is subject to frameshift introducing premature stop codons, consequently disrupting encoded functional domains.
Chimeric fusion gene transcripts ETV6-RTN1 and RTN1-ETV6. (A) In silico prediction of amino acid sequences encoded by fusion transcripts. ETV6-RTN1 fusion transcript, originating from exon 1 of ETV6 and part of exon 2 of RTN1, houses a frameshift downstream of the fusion point, resulting in a premature stop codon. The transcript is predicted to encode 86 amino acids. RTN1-ETV6 fusion transcript, originating from exon 1 of RTN1 and exons 2, 3, and part of 4 of ETV6, also houses a frameshift downstream of the fusion point resulting in a premature stop codon. This transcript is predicted to encode 178 amino acids. Electropherograms of novel exon–exon junctions, resulting from the translocation, support the in silico–predicted sequences of fusion transcripts. (B) Gel images (2% agarose gel) supporting the expression of both fusion transcripts in remission and leukemia from the 2 affected female subjects (III:3 and IV:2) and germline of an unaffected carrier (III:2). Positive control ribosomal RNA 18S5N is shown in the bottom gel image. 100bp Plus DNA Ladder from GeneRuler (Thermo Fisher Scientific). (C) Two-dimensional models of ETV6 and RTN1 protein with approximate locations of breakpoints indicated by red bars. Functional domains are not retained in theoretical fusion proteins. Breakpoints are located upstream of functional domains, and all sequence downstream of breakpoints is subject to frameshift introducing premature stop codons, consequently disrupting encoded functional domains.
Germline WGS data were also thoroughly analyzed for pathogenic single nucleotide variants and structural variants, and genes known to predispose to hematological malignancies50,,-53 were especially considered. No shared or unique pathogenic variants other than the t(12;14)(p13.2;q23.1) were found in the 2 affected female subjects or in the unaffected carrier.
Genetic analysis of somatic aberrations
The routine karyotyping analysis, performed on diagnostic bone marrow samples from the affected individuals (III:3 and IV:2) at the respective time of leukemia diagnosis, revealed no somatic aberrations in addition to the germline translocation. However, FISH analyses on the diagnostic bone marrow sample of one individual (IV:2) revealed 1 to 2 extra RUNX1 signals in 74% of the interphase nuclei. In addition, the nature of the germline karyotype t(12;14) causes 3 signals of ETV6 (1 normal and 2 split signals) on the derivative chromosomes. FISH detected 2 to 3 extra chromosome 12 signals in 48% of interphase nuclei, indicating that there is 1 or 2 extra copies of derivative chromosomes. In the scope of this study, we performed SNP arrays on genomic DNA, from the same diagnostic bone marrow samples as used for routine cytogenetic analyses at diagnosis, which revealed HeH patterns in both leukemias (Table 1); this finding is consistent with primary hyperdiploid clones. Apart from whole chromosome CNA corresponding to HeH karyotypes, as well as a partial increase in the copy number of ETV6 and RTN1 corresponding to derivative chromosomes, SNP arrays did not reveal any other somatic CNAs of genes relevant to leukemogenesis. Furthermore, coding regions of ETV6 analyzed by using Sanger sequencing did not contain any somatic single nucleotide variants or indels. Subsequently, WGS analysis of genomic DNA from diagnostic bone marrow revealed 10 and 7 somatic aberrations, respectively, in BCP-ALLs of the affected female subjects (II:3 and IV:2) (supplemental Table 3). Of the 17 genes found somatically mutated here, 13 were previously reported with somatic mutations in childhood ALLs by the Pediatric Cancer Data Portal (St. Jude Children’s Research Hospital, Memphis, TN). In addition, 2 of the variants (1 in each of the affected female subjects) were previously reported in sporadic cases of childhood ALL: the 27 kb WAC-AS1 deletion in the proband (IV:2) and the NRAS hotspot mutation p.G12S in the maternal aunt (III:3) (Pediatric Cancer Data Portal, St. Jude Children's Research Hospital [2015-2018]; https://pecan.stjude.cloud/home, accessed 20 July 2018).
Expression analysis
To evaluate the effect of the t(12;14)(p13.2;q23.1) on the expression of wild-type ETV6 and RTN1, qPCR expression analysis was performed as described in the Subject and methods section. Primers were carefully designed to only allow binding of wild-type transcripts. Germline/remission expression of wild-type ETV6 in peripheral blood showed a contradictory pattern with decreased expression in the proband (IV:2) but increased expression in the maternal aunt (III:3) (Figure 4A). In the unaffected carrier (III:2, mother of the proband), however, the expression level was similar to normal control subjects. A corresponding pattern was observed also for the expression of wild-type RTN1 in germline/remission (Figure 4B).
![Expression of wild-type ETV6 and wild-type RTN1. Expression analyzed by qPCR using primers exclusively targeting wild-type messenger RNA sequence for each gene. (A-B) Expression of wild-type ETV6 and RTN1 in germline/remission samples presented as “fold of control” [(Etarget)ΔCPtarget(control-sample)]/[(Ereference)ΔCPreference(control-sample)]. The control value is calculated as an average of all 6 normal control samples analyzed and has been given the relative value of 1 (y = 1). (C-D) Expression of wild-type ETV6 and RTN1 in leukemic bone marrow presented as ΔCt (Ct[reference] – Ct[target]). In panel D, t(12;21) is not included due to lack of material for analysis.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/18/10.1182_bloodadvances.2018028795/2/m_advances028795f4.png?Expires=1765453079&Signature=bGhyHqilIMOyRP0TNpMjHOj3RCBJG9p2utLtO6W6I2xRRAV8W531jdQCxRpH0bPK2fyjRW66XdbIFm8F7phtZEqFxxTrBHGdOeEnD07i16jhdbhGaaYkCdPuMV0BKMSpw6MV4B62fFEcu2WfnxOCV737tW8Y3TVFgYw-okiwEvbEqrsKtjVs2qtRZMIF5d0gB5KD1G7k7ojHXRkNRLpKxdfvUmrSyT4T4T8sCYXVXROnksJM5S83p1dP4cg~66JSQQ3YfdKOuBSXFn7MLo2dagrSrZNoYwhjU7OLNRY51VWEqBcBwOiNXrWI9v8fSd659FvTa7DWxMMAcPJGyLNgzA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Expression of wild-type ETV6 and wild-type RTN1. Expression analyzed by qPCR using primers exclusively targeting wild-type messenger RNA sequence for each gene. (A-B) Expression of wild-type ETV6 and RTN1 in germline/remission samples presented as “fold of control” [(Etarget)ΔCPtarget(control-sample)]/[(Ereference)ΔCPreference(control-sample)]. The control value is calculated as an average of all 6 normal control samples analyzed and has been given the relative value of 1 (y = 1). (C-D) Expression of wild-type ETV6 and RTN1 in leukemic bone marrow presented as ΔCt (Ct[reference] – Ct[target]). In panel D, t(12;21) is not included due to lack of material for analysis.
Expression of wild-type ETV6 and wild-type RTN1. Expression analyzed by qPCR using primers exclusively targeting wild-type messenger RNA sequence for each gene. (A-B) Expression of wild-type ETV6 and RTN1 in germline/remission samples presented as “fold of control” [(Etarget)ΔCPtarget(control-sample)]/[(Ereference)ΔCPreference(control-sample)]. The control value is calculated as an average of all 6 normal control samples analyzed and has been given the relative value of 1 (y = 1). (C-D) Expression of wild-type ETV6 and RTN1 in leukemic bone marrow presented as ΔCt (Ct[reference] – Ct[target]). In panel D, t(12;21) is not included due to lack of material for analysis.
Furthermore, analysis of wild-type ETV6 in leukemias showed that the affected female subjects (III:3 and IV:2) and sporadic cases with somatic heterozygous deletion of ETV6 had equivalent expression levels of wild-type ETV6 (Figure 4C). Levels were also comparable to analyzed cases with biallelic aberrations in ETV6 (t(12;21)+delETV6). However, expression levels in leukemias without ETV6 aberration overlapped all of the aforementioned cases. Moreover, the expression of wild-type RTN1 was low across all leukemic samples tested with no significant difference between the affected female subjects and sporadic control subjects (Figure 4D). Finally, expression of fusion transcripts was detected in all tested carriers (III:2, III:3, and IV:2; data not shown).
Discussion
In this study, we describe the first known family with predisposition to childhood BCP-ALL caused by a constitutional balanced reciprocal translocation disrupting ETV6. The family includes 9 confirmed carriers of t(12;14)(p13.2;q23.1); 2 of the individuals developed childhood BCP-ALL, but none displayed thrombocytopenia.
Including the family reported here, a total of 88 individuals from 23 families with pathogenic germline variants in ETV6 have been described.25,,,,,,-32 To date, 16 of 50 carriers from 11 of these families have developed ALL (15 cases confirmed BCP subtype), all displaying an autosomal dominant inheritance pattern with incomplete penetrance (supplemental Table 4).26,,-29,31,32 In addition, 38 sporadic cases of ALL (35 confirmed BCP subtype) with rare germline pathogenic variants in ETV6 have been described.28,54 A majority had a HeH karyotype (80%), older age at diagnosis (13.3 vs 6.8 years without risk variants) and leukocyte counts <50 × 109/L at diagnosis.28,33 These findings are consistent with the present study, in which 2 individuals developed HeH BCP-ALL at age 8 and 12 years, respectively, with leukocyte counts at diagnosis far below 50 × 109/L.
All previously reported families with THC5 carried pathogenic germline variants in the central domain or the DNA-binding ETS-domain of ETV6.26,,,,,,-33 A majority of these variants result in a dominant negative effect on wild-type ETV6. In contrast, the germline t(12;14)(p13.2;q23.1) studied here results in heterozygous loss of ETV6. Transcripts from the 2 actively transcribed fusion genes are most likely targeted to nonsense-mediated decay, but if translated, the resulting fusion gene products would be nonfunctional. Likewise, the only previously reported family without thrombocytopenia also had a clear heterozygous loss of ETV6, due to an early truncating variant (p.W72*).25 Interestingly, 5 other families with truncating germline variants located in the ETS-domain of ETV6 have been reported. In the 2 with more proximal variants (p.R359* and p.R378*), THC5 occurred with reduced penetrance,26,28 whereas full penetrance was observed in the 3 families with the most distal variant (N385Vfs*/R418G).27,29,31 Similarly, 62 (100%) of 62 individuals with germline missense variants in ETV6 were reported to have THC5 with full penetrance.27,29,,-32 Thus, THC5 seems to be associated with more severe functional deficiency of ETV6 caused by a dominant negative effect on wild-type protein (supplemental Table 4).
We hypothesized that haploinsufficiency of ETV6 may contribute to BCP-ALL predisposition in the studied family, but expression analyses were inconclusive and could not affirm the hypothesis in t(12;14) carriers. Tested carriers exhibited contradictive differences in germline expression of wild-type ETV6 (Figure 4A), whereas expression of wild-type ETV6 in leukemic bone marrow did not deviate substantially when comparing the affected female subjects vs sporadic case subjects wild type for (and with) various aberrations in ETV6 (Figure 4C). The normal variation of cell type composition in a sample combined with substantial differences in ETV6 expression between various cell types in peripheral blood55 could have obscured differences in wild-type ETV6 expression caused by the translocation. Therefore, haploinsufficiency cannot yet be ruled out as a mechanism of BCP-ALL predisposition in this family. Thus far, there is no support for RTN1 having a role in childhood leukemia development, either for sporadic or predisposed cases. Therefore, based on the well-known association between ETV6 and BCP-ALL and the phenotypic similarities between our case subjects and previously reported leukemias with ETV6-mediated predisposition, we believe that ETV6 is the responsible gene behind the leukemia predisposition in this family. Altogether, we suggest that monoallelic expression of ETV6 may have a sufficient effect on normal B-cell development to contribute to leukemia predisposition, warranting further investigation.
Previous reports of individuals with germline pathogenic variants in ETV6 and BCP-ALL show that additional somatic aberrations in ETV6 or other leukemia-associated genes are required for leukemia development.28,32 Likewise, healthy individuals with prenatal existence of somatic ETV6-RUNX1 fusion transcripts also require additional somatic aberrations for development of BCP-ALL.56,57 In the present study, although no somatic second hits were found in ETV6 or RTN1, a number of somatic pathogenic variants were identified in both leukemias. The majority of genes (13 of 17) found somatically mutated were previously known to be subject to somatic mutations in childhood ALL, but only 2 of the variants (NRAS missense variant and 27 Kb long deletion in WAC-AS1) were previously reported in childhood ALL (Pediatric Cancer Data Portal, St. Jude Children's Research Hospital [2015-2018]; https://pecan.stjude.cloud/home, accessed 20 July 2018). None of the detected somatic variants were found to be shared between the affected female subjects. The extra chromosomes in the HeH clones were partly overlapping but consistent with common ploidy patterns for HeH BCP-ALL in both leukemias.58 Thus, in terms of somatic variants, the leukemias studied here seem to have developed differently, despite the shared predisposing factor. The sequence of somatic events required for overt leukemia in cases with germline pathogenic variants in ETV6 remains to be understood.
Solid tumors have also been reported in families carrying pathogenic germline variants in ETV6.27,29,32,54 In addition, secondary malignancies, both hematological and solid, have been reported in childhood ALL cases with germline variants in ETV6.31,54 Even so, there is not yet compelling evidence regarding ETV6 germline predisposition to solid tumors,59 but further investigation is needed.
In conclusion, we report the first known family with predisposition to childhood BCP-ALL caused by a constitutional balanced reciprocal translocation disrupting ETV6. We suggest that germline aberrations resulting in monoallelic expression of ETV6 may contribute to leukemia susceptibility, whereas more severe functional deficiency of ETV6 is required for THC5 development. Further studies to investigate the importance of bi-allelic ETV6 expression for normal hematopoiesis are required. Additional reports of families with various germline aberrations in ETV6 would also be valuable to expand the understanding of genotype–phenotype associations. Finally, we highlight the importance of constitutional structural aberrations in childhood cancer predisposition and emphasize the advantage of WGS in understanding their molecular consequences.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the head of the hematology laboratory, Eeva-Riitta Savolainen (NordLab Oulu, Oulu University Hospital) and Kirsi Kvist-Mäkelä (NordLab Oulu, Oulu University Hospital) for patient sample preparation and DNA extraction. They are grateful to Mikko Seppänen for providing clinical data. They are also grateful to Anh-Nhi Tran, Yenan Bryceson and Heinrich Schlums for providing control samples and for fruitful discussions. The authors also thank Magnus Nordenskjöld for providing scientific expertise.
This work was supported by grants from the Swedish Childhood Cancer Foundation, the Swedish Cancer Society, the Cancer Research Funds of Radiumhemmet, the Swedish Research Council, Berth von Kantzow’s foundation, the Mary Béve Foundation for Pediatric Cancer Research, Karolinska Institutet, the Alma and K. A. Snellman Foundation, Maud Kuistila Memorial Foundation, the Finnish Foundation for Pediatric Research, and Väre Foundation for Pediatric Cancer Research.
Authorship
Contribution: T.J. and R.N. gathered and analyzed the clinical data; B.B., T.J., and V.Z. performed experiments; F.T. provided data and analysis; B.B., V.Z., F.T., and T.J. analyzed genetic data; T.J., B.B., V.Z, R.N., and A.N. wrote the manuscript; J.M., M.M., A.H.-S., and R.N. enrolled the patients in the study and provided clinical data; C.I.E.S. provided expertise on immunology; A.H.-S., R.N., and A.N. designed and supervised the research; and all authors revised and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ann Nordgren, Department of Molecular Medicine and Surgery and Centre for Molecular Medicine, Karolinska Institute, 171 76 Stockholm, Sweden; e-mail: ann.nordgren@ki.se.




![Expression of wild-type ETV6 and wild-type RTN1. Expression analyzed by qPCR using primers exclusively targeting wild-type messenger RNA sequence for each gene. (A-B) Expression of wild-type ETV6 and RTN1 in germline/remission samples presented as “fold of control” [(Etarget)ΔCPtarget(control-sample)]/[(Ereference)ΔCPreference(control-sample)]. The control value is calculated as an average of all 6 normal control samples analyzed and has been given the relative value of 1 (y = 1). (C-D) Expression of wild-type ETV6 and RTN1 in leukemic bone marrow presented as ΔCt (Ct[reference] – Ct[target]). In panel D, t(12;21) is not included due to lack of material for analysis.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/18/10.1182_bloodadvances.2018028795/2/m_advances028795f4.png?Expires=1765453080&Signature=cU-yvL4N8-qPs4NsKepVTFoAGeUUCHqSxXA4RUgJSR6gtFLegU2yvSc7EWKP1yEYO~~JUQB2SlVInlM32D5WqWkn80g1memALNd4f3CsJwRfRroi9c3473ZX1AHYTfHzQztRkdydJ7h9xjcRokBeKkkjlyvxYmscH~H1jfzkw0OjwGaJ-eIC3B2t47Aqo0qztJjSzc9RptSDRBABETlb4~QrOJcy~mxM7IRqJVdhZBl10X89X~DwIQVErymVwdiGL9hY-AeymJLXVCw1vkOkg~RrwORJTakZbgvVg8ohZV-lmOgsQxMVqyaJ9zPlX68xmy~Xo3o~jvPm3CG3LuCgxQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)

