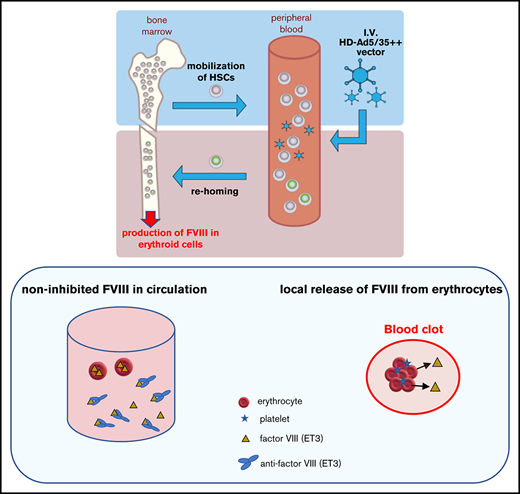
Key Points
An in vivo HSC transduction/selection allows for high-level protein expression from erythroid cells without side effects on erythropoiesis.
This approach that did not require ex vivo HSC manipulation and transplantation resulted in phenotypic correction of murine hemophilia A.

Abstract
We developed an in vivo hematopoietic stem cell (HSC) transduction approach that involves HSC mobilization from the bone marrow into the peripheral bloodstream and the IV injection of an integrating, helper-dependent adenovirus (HDAd5/35++) vector system. HDAd5/35++ vectors target human CD46, a receptor that is abundantly expressed on primitive HSCs. Transgene integration is achieved by a hyperactive Sleeping Beauty transposase (SB100x) and transgene marking in peripheral blood cells can be increased by in vivo selection. Here we directed transgene expression to HSC-derived erythroid cells using β-globin regulatory elements. We hypothesized that the abundance and systemic distribution of erythroid cells can be harnessed for high-level production of therapeutic proteins. We first demonstrated that our approach allowed for sustained, erythroid-lineage specific GFP expression and accumulation of GFP protein in erythrocytes. Furthermore, after in vivo HSC transduction/selection in hCD46-transgenic mice, we demonstrated stable supraphysiological plasma concentrations of a bioengineered human factor VIII, termed ET3. High-level ET3 production in erythroid cells did not affect erythropoiesis. A phenotypic correction of bleeding was observed after in vivo HSC transduction of hCD46+/+/F8−/− hemophilia A mice despite high plasma anti-ET3 antibody titers. This suggests that ET3 levels were high enough to provide sufficient noninhibited ET3 systemically and/or locally (in blood clots) to control bleeding. In addition to its relevance for hemophilia A gene therapy, our approach has implications for the therapy of other inherited or acquired diseases that require high levels of therapeutic proteins in the blood circulation.
Introduction
Current hematopoietic stem cell (HSC) gene therapy protocols are complex, involving the collection of HSCs from donors/patients by leukapheresis, in vitro culture, transduction with lentivirus vectors, and retransplantation into myeloconditioned patients. Besides the technical complexity, the cost of the approach prohibits a widespread application. We developed a minimally invasive and readily translatable approach for in vivo HSC gene delivery without leukapheresis, myeloablation, and HSC transplantation. We showed that in vivo transduction of primitive HSCs is safe and efficient using a simple procedure that involves HSC mobilization with standard drugs (granulocyte colony-stimulating factor [G-CSF]/AMD3100) and IV injection of hCD46-targeting helper-dependent adenovirus (HDAd5/35++) vectors. HDAd5/35++ vectors are helper-dependent vectors devoid of all viral genes and containing modified Ad serotype 35 fibers that detarget the vector from the liver and allow for efficient HSC transduction. HSCs, transduced in the periphery, return to the bone marrow.1 Stable HSC genome modification in mice can be achieved by integrating HDAd5/35++ vectors using a hyperactive Sleeping Beauty transposase (SB100x).2,3 Without a disease-related preferential survival bias, mgmtP140K expression and low-dose treatment with O6BG/BCNU (“in vivo selection”) is required to achieve efficient (90% to 100%) transgene marking in peripheral blood cells.4 Using a human γ-globin gene under control of a mini-β-globin locus control regions (LCR), the in vivo HSC transduction/selection approach achieved near complete correction in a mouse model of thalassemia intermedia.5 Here, we explored the possibility of whether our approach can be employed for the production of nonerythroid proteins in erythroid lineage cells and whether it would phenotypically correct hemophilia A in mice.
Approximately 2.4 million new erythrocytes are produced per second in human adults. Nearly a quarter of the cells in the human body are red blood cells (RBCs).6 In the final stages of erythropoiesis, HSCs differentiate through common myeloid progenitors and preerythroblasts to orthochromatic erythroblasts (based on Wright’s stain). At this stage, the nucleus is expelled, and the cells exit the bone marrow into the circulation as reticulocytes. About 0.5% to 2.5% of circulating RBCs in adults (∼1 × 105/μL) and 2% to 6% in infants are reticulocytes. Reticulocytes still produce hemoglobin from messenger RNA (mRNA). After 1 to 2 days, these cells ultimately lose all organelles and become mature RBCs, which are not capable of protein biosynthesis anymore. Differentiation from committed erythroid progenitors to erythrocytes takes ∼7 days. Erythrocytes release their contents after senescence. Old and dying erythrocytes are removed by the phagocytic system of the spleen. Once HSCs have differentiated into committed erythroid cells, enormous amounts of α- and β-globin chains are produced and then later stored in erythrocytes as tetrameric hemoglobin. A healthy individual has 12 to 20 g of hemoglobin per 100 mL of blood, and ∼95% of the erythrocyte weight is hemoglobin (270 × 106 hemoglobin molecules per cell). The basis for this efficient biosynthesis is strong erythroid-specific LCRs that allow for high-level transcription and stable mRNA that is efficiently translated.
We capitalized on the tremendous speed and efficacy of erythropoiesis and the powerful machinery for hemoglobin synthesis to produce nonerythroid proteins from erythrocyte precursor cells (encompassing the differentiation stages from proerythroblasts to reticulocytes). Transgenes were under the control of a mini-β-globin LCR and contained 3′ untranslated region (UTR) of the β-globin gene for mRNA stabilization. Furthermore, we hypothesized that proteins are released from mature erythrocytes during/after senescence, thus contributing to locally and systemically high protein levels in blood.
As a proof-of-principle that erythroid cells can be used for high-level production of therapeutic proteins that are released into the blood circulation, we focused here on coagulation factor VIII. The outcome of the study is relevant for hemophilia A treatment. Near complete correction of hemophilia A has now been achieved in patients by hepatic in vivo gene transfer using recombinant AAV vectors.7 However, the widespread application of liver-directed rAAV hemophilia A gene therapy could face several obstacles: (1) mostly episomal nature of rAAV genomes in hepatocytes and their loss due to cell division, specifically in children; (2) the high cost of rAAV vector production, estimated to be >$1 000 000 per patient; (3) the limited packaging capacity of rAAV that cannot accommodate large transcriptionally regulatory elements often required to prevent gene silencing or genotoxicity8,9 ; and (4) the increased risk of tumorigenicity due to potential rAAV integration near protooncogenes,10 specifically in patients with underlying liver disease, such as viral hepatitis, or in children with actively dividing hepatocytes, which represent a large fraction of hemophilia patients.11,12 Our approach to express FVIII from erythroid cells using integrating HDAd5/35++ vectors could address these problems.
Progress has been made by bioengineering of the FVIII protein to overcome its inefficient biosynthesis and secretion.13 We used an FVIII mutant designated ET3 that had been shown to confer 10- to 100-fold improved biosynthesis.14 The ET3 complementary DNA (cDNA) (∼4.5 kb) is deleted for the B-domain and has porcine amino-acid substitutions in the A1 and ap-A3 domains of human FVIII. The ET3 amino acid sequence has 91% identity to the B-domain deleted human FVIII.15 Liver-directed rAAV gene transfer of the ET3 transgene into hemophilia A mice enabled high-level expression at low vector dose and long-term correction of bleeding.14 More recently, an ex vivo HSC gene therapy approach has been developed where the ET3 gene is expressed from a CD68 promoter.16,,-19
In our study, we show, using GFP as a reporter gene under control of the β-globin mini-LCR, that after in vivo HSC transduction/selection it is possible to achieve expression of a nonerythroid protein in erythroid cells and storage of GFP in mature RBCs. We then demonstrate that our approach results in supraphysiological levels of ET3 and phenotypic correction in a hemophilia A mouse model despite the presence of high-titer anti-ET3 plasma antibodies.
Materials and methods
Reagents
The following reagents were used: G-CSF (Neupogen; Amgen, Thousand Oaks, CA), AMD3100 (Sigma-Aldrich, St. Louis, MO), Plerixafor (Mozobil; Genzyme Corp, Cambridge, MA), O6-BG and BCNU (Sigma-Aldrich), mycophenolate mofetil (CellCept Intravenous; Genentech, Hillsboro, OR), rapamycin (Rapamune/Sirolimus; Pfizer, New York, NY), methylprednisolone (Pfizer).
Generation of HDAd vectors
The generation of HDAd-LCR-ET3/mgmt and HDAd-LCR-GFP/mgmt is described in supplemental Methods. For the production of HDAd5/35++ vectors, corresponding plasmids were linearized with FseI and rescued in 116 cells20 with Ad5/35++-Acr helper vector2 as described in detail elsewhere.20 Helper virus contamination levels were found to be <0.05%. Titers were 6 to 12 × 1012 vp/mL.
All HDAd vectors used in this study contain chimeric fibers composed of the Ad5 fiber tail, the Ad35 fiber shaft, and the affinity-enhanced Ad35++ fiber knob.21
Cells
HUDEP-2 cells22 were provided by Ryo Kurita (Department of Research and Development, Central Blood Institute, Japanese Red Cross Society, Tokyo, Japan) and Yukio Nakamura (Cell Engineering Division, RIKEN BioSource Center, Ibaraki, Japan). HUDEP-2 cells were cultured in the presence of stem cell factor, erythropoietin, doxycyclin, and dexamethasone as previously described.23 The cells were transduced with the HDAd vectors at a multiplicity of infection of 500 to 1000 vp per cell and analyzed as indicated.
Animal studies
The hemophilia A mice (B6;129S-F8tm1Kaz/J) were purchased from Jackson Laboratories. These mice were crossed with hCD46+/+ transgenic mice to obtain hCD46+/−/ F8−/−mice for ex vivo studies and hCD46+/+/ F8−/−double transgenic mice for in vivo HSC transduction studies. The following primers were used for genotyping of hCD46 mice: forward, 5′-GCCAGTTCATCTTTTGACTCTATTAA-3′, and reverse, 5′-AATCACAGCAATGACCCAAA-3′. Mice homozygous or heterozygous for hCD46 were identified by different intensity of hCD46 expression on peripheral blood mononuclear cells detected by flow cytometry. Genotyping of mouse factor VIII gene disruption was performed by polymerase chain reaction (PCR) according to Jackson Labs’ recommended primers and protocol.
Bone marrow Lin− cell transplantation.
Recipients were hemophilia A (B6;129S-F8tm1Kaz/J) mice, 6 to 8 weeks old. On the day of transplantation, recipient mice were irradiated with 1000 Rad. Four hours after irradiation, 0.5 to 1 × 106 Lin− cells were injected IV through retroorbital injection. This protocol was used for transplantation of ex vivo transduction Lin− cells and for transplantation into secondary recipients.
HSC mobilization and in vivo transduction.
This procedure was described previously.24 Briefly, HSCs were mobilized in mice by subcutaneous injections of human recombinant G-CSF (5 μg per mouse per day, 4 days) (Amgen) followed by a subcutaneous injection of AMD3100 (5 mg/kg) (Sigma-Aldrich) on day 5. In addition, animals received dexamethasone (10 mg/kg) intraperitoneally (IP) 16 hours and 2 hours before virus injection. Thirty and 60 minutes after AMD3100, animals were IV injected with HDAd-LCR-ET3/mgmt or HDAd-LCR-GFP/mgmt and HDAd-SB through the retroorbital plexus with a dose of 4 × 1010 vp for each virus per injection. Following treatment, combined immunosuppression was administered. At week 4, mice were subjected to 4 cycles of in vivo selection with O6BG (30 mg/kg, IP) and escalated BCNU doses (5, 7.5, 10, 10 mg/kg) with a 2-week interval between doses. Immunosuppression was resumed 2 weeks after the last O6-BG/BCNU dose. In mice in which bleeding could not be stopped after IV injections, 500 to 1000 ng recombinant ET3 protein was injected IP.
Immunosuppression.
Daily IP injection of mycophenolate mofetil (20 mg/kg per day), rapamycin (0.2 mg/kg per day), and methylprednisolone (20 mg/kg per day) was performed.
Tail bleeding assay
Mice were anesthetized under isoflorane and were fixed in the supine position on a temperature (37°C)-controlled heating table, and their tails were warmed for 10 minutes in a 37°C water bath. Tails were transected at a distance of 10 mm from the tip using a surgical blade and immediately placed into 15-mL tubes containing 14 mL of 0.9% saline solution held at 37°C. Tails were allowed to bleed freely for 45 minutes, at which time they were removed from the vials. Blood cells were collected by centrifugation at 1500 rpm/min for 10 minutes at room temperature. Cells (the vast majority of which were RBCs) were resuspended in 10 mL of Drabkin’s solution (Sigma), vortexed, and allowed to stand for at least 15 minutes at room temperature. Concentrations of hemoglobin were measured spectrophotometrically at 540 nm and determined using the absorbance of cyanmet-hemoglobin standard solutions (Hemoglobin human; Sigma). The total hemoglobin loss was calculated and normalized to the body weight (measured before the experiment).
Plasma antibodies against ET3
The assay has been described previously.25 Briefly, recombinant ET3 protein (5 μg/mL in phosphate-buffered saline) was immobilized on 96-well enzyme-linked immunosorbent assay (ELISA) plates overnight at 4°C. After blocking with StartingBlock blocking buffer (Thermo Scientific), serially diluted mouse plasma was added to the wells. Anti-ET3 antibodies were detected using a goat–anti-mouse immunoglobulin G (IgG) secondary antibody conjugated with horseradish peroxidase (BD Pharmingen). Binding was detected as described for the ET3 ELISA. The absorbance values of test plasmas were plotted against the logarithm of the plasma dilution, and the titers were determined to be the reciprocal of the dilution in which the optical density value was 3 times that of the background (the original hemophilia A mice [B6;129S-F8tm1Kaz/J] plasma), or an optical density reading of 0.3.
Thrombin/antithrombin assay
Thrombin/antithrombin complex levels in mouse plasma were measured by mouse thrombin-antithrombin complexes ELISA kit (Abcam) following manufacturer’s instructions.
Statistical analyses
For comparisons of multiple groups, 1-way and 2-way analysis of variance (ANOVA) with Bonferroni posttesting for multiple comparisons was employed. Statistical analysis was performed using GraphPad Prism version 6.01 (GraphPad Software Inc, La Jolla, CA). For nonparametric analyses, the Kruskal-Wallis test was used.
The supplemental Methods section contains the description for generation of HDAd vectors, approval information for animal studies, isolation of lineage-depleted (Lin−) bone marrow cells, colony-forming unit (CFU) assay, real-time reverse transcription PCR for ET3 mRNA, measurement of vector copy number (VCN), ELISA for plasma ET3, chromogenic activity assay for plasma ET3, ET3 western blot, VCN in single colonies, GFP immunofluorescence analysis on tissue slides, hematological analyses, and flow cytometry.
Results
Expression of GFP in erythroid cells after in vivo HSC transduction of hCD46 transgenic mice
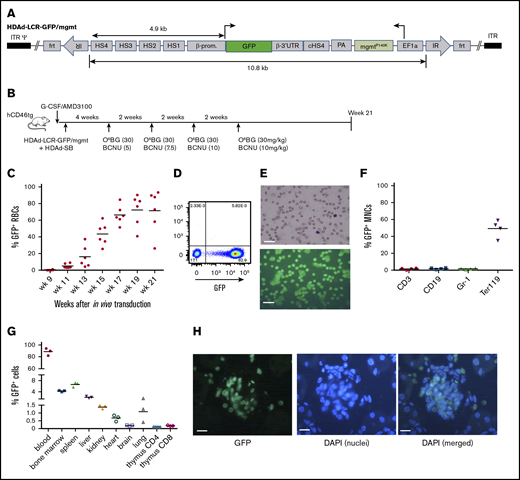
As a proof-of-principle that erythroid cells can express nonerythroid proteins, we generated an HDAd5/35++ vector containing the GFP gene under the control of a β-globin mini-LCR (HDAd-LCR-GFP/mgtm) (Figure 1A). In previous studies, this LCR version conferred high-level erythroid-specific expression of globin genes after ex vivo or in vivo HSC transduction.5,26 The vector also contained a mgmtP140K gene under the control of the ubiquitously active EF1α promoter, which allowed for in vivo selection of transduced progenitors with O6BG/BCNU and controlled increase of transgene marking in erythroid cells. For in vivo HSC transduction, hCD46-transgenic mice were mobilized and IV injected with HDAd-LCR-GFP/mgtm plus HDAd-SB followed by 4 cycles of O6BG/BCNU treatment (at weeks 4, 6, 8, and 10 after HDAd injection) (Figure 1B). As a result of in vivo selection, GFP marking in peripheral RBCs increased to an average of 80% and remained stable until the end of the study (week 21 after in vivo transduction) (Figure 1C). Remarkable was the high GFP fluorescence intensity of erythrocytes (Figure 1D-E). Biodistribution of GFP-positive cells was analyzed at week 21. In the bone marrow, ∼50% of erythroid (Terr119+) cells were GFP positive (Figure 1F). The percentage of GFP+ cells in the nonerythroid lineages (CD3, CD19, and Gr-1) was ∼30-fold lower. In the spleen and liver, ∼6 and ∼2.5% GFP+ cells, respectively, were found (Figure 1G; supplemental Figure 1). Less than 1.5% GFP+ cells were present in kidney, heart, brain, and lung. In the thymus, ∼0.1% and ∼0.2% of CD4 and CD8 T cells, respectively, expressed the GFP transgene. Analysis of tissue sections revealed that most GFP+ cells in tissues were erythrocytes inside blood vessels, as shown for kidney (Figure 1H) or liver (supplemental Figure 2). Despite high-level GFP expression/accumulation in erythroid cells, no hematological abnormalities were observed in mice at week 21 (supplemental Figure 3).
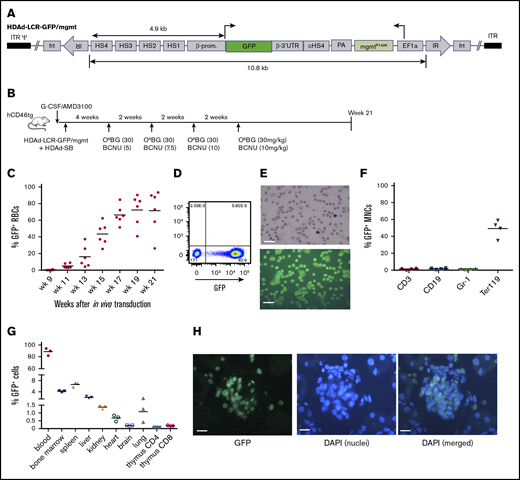
GFP expression in erythroid cells after in vivo HSC transduction/selection. (A) HDAd-LCR-GFP/mgmt vector for SB100x transposase-mediated integration. The GFP gene is under the control of a 4.9-kb erythroid-specific (human) β-globin “mini” LCR consisting of DNase I hypersensitivity regions (HS) 1 to 4 and the β-globin promoter. The GFP gene is linked to the β-globin 3′UTR for mRNA stabilization in erythroid cells. The ET3 and mgmt expression cassettes are separated by a chicken globin HS4 insulator. The 10.8-kb ET3/mgmt transposon is flanked by inverted repeats (IRs) that are recognized by SB100x, mediating random integration. (B) In vivo transduction of mobilized hCD46-transgenic mice. HSCs were mobilized, and animals were IV injected with a 1:1 mixture of HDAd-LCR-ET3/mgmt + HDAd-SB. At week 4, O6-BG/BCNU treatment was started and repeated every 2 weeks 4 times. With each cycle, the BCNU concentration was increased from 5 mg/kg, to 7.5 mg/kg, to 10 mg/kg. (C) Percentage of GFP-positive peripheral RBCs. Each symbol is an individual animal. (D) Representative flow cytometry analysis for GFP expression in total blood cells at week 21 after in vivo HSC transduction. (E) Peripheral blood smears were stained with May-Grünwald Giemsa (Merck) for 5 minutes (upper panel). GFP fluorescence analyzed on week 21 blood smears (lower panel). Scale bars, 20 μm. (F) Percentage of GFP-positive mononuclear cells (MNCs) measured by flow cytometry in different lineages in the bone marrow. CD3, T-lymphocytes/progenitors; CD19, B-lymphocytes/progenitors; Gr-1, granulocytes/progenitors; Ter119, erythroid cells. (G) Percentage of GFP-positive cells in different tissues measured by flow cytometry of single-cell suspensions. Note that the y-axis has 3 segments. (H) Representative kidney section demonstrating the presence of GFP-positive erythrocytes in a glomerulus. 4′,6-Diamidino-2-phenylindole (DAPI) (blue) was used to visualize cell nuclei. In vivo HSC transduced mice were kept alive without side effects for 21 weeks. Scale bars, 20 μm.
GFP expression in erythroid cells after in vivo HSC transduction/selection. (A) HDAd-LCR-GFP/mgmt vector for SB100x transposase-mediated integration. The GFP gene is under the control of a 4.9-kb erythroid-specific (human) β-globin “mini” LCR consisting of DNase I hypersensitivity regions (HS) 1 to 4 and the β-globin promoter. The GFP gene is linked to the β-globin 3′UTR for mRNA stabilization in erythroid cells. The ET3 and mgmt expression cassettes are separated by a chicken globin HS4 insulator. The 10.8-kb ET3/mgmt transposon is flanked by inverted repeats (IRs) that are recognized by SB100x, mediating random integration. (B) In vivo transduction of mobilized hCD46-transgenic mice. HSCs were mobilized, and animals were IV injected with a 1:1 mixture of HDAd-LCR-ET3/mgmt + HDAd-SB. At week 4, O6-BG/BCNU treatment was started and repeated every 2 weeks 4 times. With each cycle, the BCNU concentration was increased from 5 mg/kg, to 7.5 mg/kg, to 10 mg/kg. (C) Percentage of GFP-positive peripheral RBCs. Each symbol is an individual animal. (D) Representative flow cytometry analysis for GFP expression in total blood cells at week 21 after in vivo HSC transduction. (E) Peripheral blood smears were stained with May-Grünwald Giemsa (Merck) for 5 minutes (upper panel). GFP fluorescence analyzed on week 21 blood smears (lower panel). Scale bars, 20 μm. (F) Percentage of GFP-positive mononuclear cells (MNCs) measured by flow cytometry in different lineages in the bone marrow. CD3, T-lymphocytes/progenitors; CD19, B-lymphocytes/progenitors; Gr-1, granulocytes/progenitors; Ter119, erythroid cells. (G) Percentage of GFP-positive cells in different tissues measured by flow cytometry of single-cell suspensions. Note that the y-axis has 3 segments. (H) Representative kidney section demonstrating the presence of GFP-positive erythrocytes in a glomerulus. 4′,6-Diamidino-2-phenylindole (DAPI) (blue) was used to visualize cell nuclei. In vivo HSC transduced mice were kept alive without side effects for 21 weeks. Scale bars, 20 μm.
Our data demonstrate strong erythroid lineage-specificity of GFP expression and accumulation of GFP protein in erythrocytes without side effects.
In vivo HSC transduction with HDAd-LCR-ET3/mgmt in hCD46-transgenic “healthy” mice
To test the concept of expressing a secreted therapeutic transgene product from erythrocytes, we generated an HDAd5/35++ vector containing the ET3 gene under control of the mini-LCR (Figure 1A) (HDAd-LCR-ET3/mgmt). The use of a protein that can be detected in plasma should also allow for studying the kinetics of transgene expression upon in vivo HSC transduction/selection.
A first in vitro study was performed in HUDEP-2 cells, an immortalized human umbilical cord blood–derived erythroid progenitor cell line, which can be differentiated into mature erythroid cells. After transduction with HDAd-LCR-ET3/mgmt plus HDAd-SB and subsequent erythroid differentiation, ET3/FVIII activity in the culture supernatant was ∼70% of that of human plasma (supplemental Figure 4A-B). Western blot analysis showed a product of 168 kDa, which is the theoretic molecular weight of single-chain ET3 (supplemental Figure 4C).
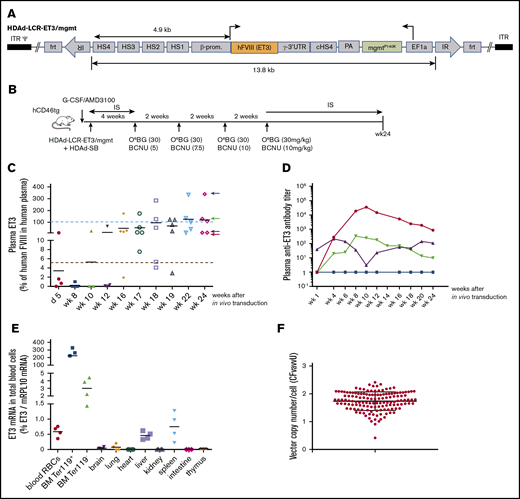
After demonstrating that our vector system produced functional ET3, we performed an in vivo HSC transduction/selection study in hCD46-transgenic mice (Figure 2B). These mice have normal plasma factor VIII activity. Using an ELISA with monoclonal antibodies capable of distinguishing mouse and human factor VIII, we measured plasma ET3 concentrations over 24 weeks (Figure 2C). ET3 detected at day 5 after in vivo HSC transduction most likely originated from episomal vectors. By week 8, plasma ET3 levels declined, likely due to loss of episomal HDAd vector copies. After completion of in vivo selection (week 10), ET3 levels steadily increased reaching supraphysiological levels in 3 out of 5 mice at week 24 (physiological level: 100% = 1 U/mL = 2180 ng/mL of ET3). The plasma ET3 level in 1 animal was ∼7.5 μg/mL (Figure 2C blue arrow). This animal did not develop anti-ET3 plasma antibodies (Figure 2D). In other animals, after an initial rise, antibody levels declined, indicating some degree of tolerance induction. ET3 mRNA levels were measured by quantitative reverse transcription polymerase chain reaction (qRT-PCR) in total blood cells (the vast majority of which are RBCs) (Figure 2E first panel) as well as in erythroid (Ter119+) and nonerythroid (Ter119−) bone marrow fractions (Figure 2E second and third panels). As expected, we found ∼400-fold higher ET3 mRNA levels in bone marrow Ter119+ cells than in peripheral blood RBCs. In the bone marrow, levels in Ter119+ cells were ∼80-fold higher than in nonerythroid cells. ET3 mRNA levels in tissues other than liver and spleen were barely above detection limit.
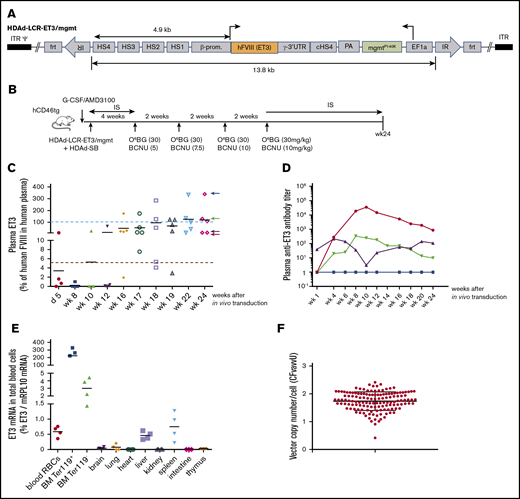
In vivo HSC transduction with HDAd-LCR-ET3/mgmt in hCD46-transgenic “healthy” mice. (A) Vector structure. See description of Figure 1A. The HDAd-LCR-ET3/mgmt vector contains the cDNA of ET3 instead of the GFP gene. (B) The treatment regimen is the same as shown in Figure 1B. Instead of HDAd-LCR-GFP, the factor VIII/ET3 expressing vector HDAd-LCR-ET3/mgtm was used. (C) Plasma concentrations of ET3 measured by ELISA. Each symbol is an individual animal. The values are expressed relative to FVIII levels in human plasma. Therefore, 100% (= 2180 ng/mL) would correspond to 1 U/mL. The dotted red line indicates 5% of physiological levels, which would correspond to a mild hemophilia A in patients and is therefore considered a therapeutic target.7 (D) Representative profiles of plasma anti-ET3 antibodies. Animals fall into the 4 groups indicated by corresponding colored arrows on the right side of the last panel in C. (E) ET3 mRNA levels measured by qRT-PCR relative to mouse mRPL10 mRNA levels. (F) Integrated ET3 cDNA copies per cell (VCN) in CFU. Bone marrow Lin− cells (pooled from 3 mice) were isolated at week 24 and plated for CFU assay. Individual colonies were picked, and the VCN was measured. Each symbol is an individual colony. In vivo HSC transduced mice were kept alive without side effects for 24 weeks.
In vivo HSC transduction with HDAd-LCR-ET3/mgmt in hCD46-transgenic “healthy” mice. (A) Vector structure. See description of Figure 1A. The HDAd-LCR-ET3/mgmt vector contains the cDNA of ET3 instead of the GFP gene. (B) The treatment regimen is the same as shown in Figure 1B. Instead of HDAd-LCR-GFP, the factor VIII/ET3 expressing vector HDAd-LCR-ET3/mgtm was used. (C) Plasma concentrations of ET3 measured by ELISA. Each symbol is an individual animal. The values are expressed relative to FVIII levels in human plasma. Therefore, 100% (= 2180 ng/mL) would correspond to 1 U/mL. The dotted red line indicates 5% of physiological levels, which would correspond to a mild hemophilia A in patients and is therefore considered a therapeutic target.7 (D) Representative profiles of plasma anti-ET3 antibodies. Animals fall into the 4 groups indicated by corresponding colored arrows on the right side of the last panel in C. (E) ET3 mRNA levels measured by qRT-PCR relative to mouse mRPL10 mRNA levels. (F) Integrated ET3 cDNA copies per cell (VCN) in CFU. Bone marrow Lin− cells (pooled from 3 mice) were isolated at week 24 and plated for CFU assay. Individual colonies were picked, and the VCN was measured. Each symbol is an individual colony. In vivo HSC transduced mice were kept alive without side effects for 24 weeks.
We also attempted to measure the integrated transgene copy number (VCN) at a single-cell level. To do this, we plated bone marrow Lin− cells collected from 4 mice at week 24 (see Figure 2C) for progenitor CFU assays, picked single colonies, and performed quantitative PCR with ET3-specific primers (Figure 2F). Remarkably, all CFU contained integrated vector DNA with an average VCN of ∼2 copies per cell. This underscores the efficiency of our in vivo HSC transduction/selection approach.
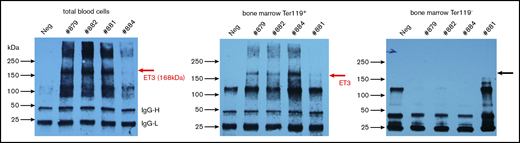
To visualize de novo produced ET3 protein, we performed an immunoprecipitation/western blot study. A band at 168 kDa was present in lysates of total blood cells/RBCs and bone marrow Ter119+ cells but was not detected in nonerythroid bone marrow cells (Figure 3). Despite high-level ET3 production in erythroid cells, no hematological abnormalities were found (Figure 4A-C). The percentage of lineage-positive cell fractions, specifically Ter119+ cells, was comparable in mice before treatment and at week 24 after in vivo HSC transduction indicating no detrimental effects on erythropoiesis (Figure 4D).

Erythroid-specific expression of ET3. Immunoprecipitation/western blot with lysates from total blood cells (ie, mostly erythrocytes) and erythroid (Ter119+) and nonerythroid (Ter119−) bone marrow fractions. Each lane is an individual mouse. The specific ET3 band appears ∼168 kDa. IgG-H and IgG-L labels bands for light and heavy immunoglobulin chains.
Erythroid-specific expression of ET3. Immunoprecipitation/western blot with lysates from total blood cells (ie, mostly erythrocytes) and erythroid (Ter119+) and nonerythroid (Ter119−) bone marrow fractions. Each lane is an individual mouse. The specific ET3 band appears ∼168 kDa. IgG-H and IgG-L labels bands for light and heavy immunoglobulin chains.
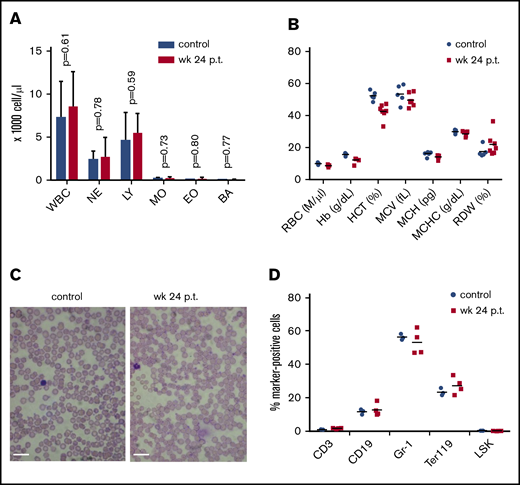
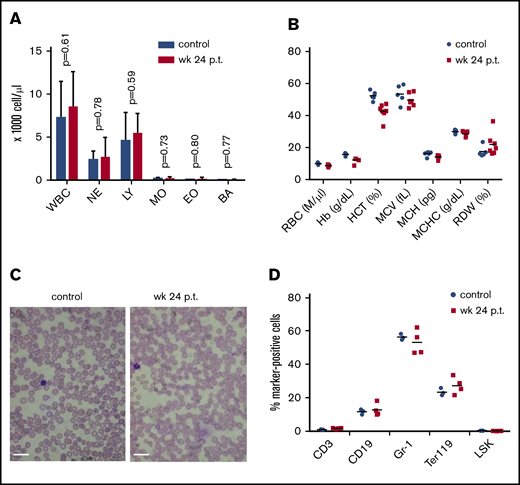
Effect of high-level ET3 expression on erythropoiesis. (A) Blood cell counts in hCD46-transgenic mice before treatment and at week 24 after in vivo HSC transduction. (B) Hematological parameters. Statistical analysis was performed using 2-way ANOVA. The differences between the 2 groups were not significant. (C) Representative blood smears before treatment and at week 24 after treatment in a mouse that expressed high-level ET3. Nuclei of white blood cells (WBCs) stain purple. Scale bars, 20 μm. (D) Cellular bone marrow composition in control and treated mice euthanized at week 24. Shown is the percentage of lineage marker-positive cells (Ter119+, CD3+, CD19+, and Gr-1+ cells). The differences between the 2 groups were not significant. BA, basophils; EO, eosinophils; Hb, hemoglobin; HCT, hematocrit; LY, lymphocytes; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; NE, neutrophils; RDW, red cell distribution width.
Effect of high-level ET3 expression on erythropoiesis. (A) Blood cell counts in hCD46-transgenic mice before treatment and at week 24 after in vivo HSC transduction. (B) Hematological parameters. Statistical analysis was performed using 2-way ANOVA. The differences between the 2 groups were not significant. (C) Representative blood smears before treatment and at week 24 after treatment in a mouse that expressed high-level ET3. Nuclei of white blood cells (WBCs) stain purple. Scale bars, 20 μm. (D) Cellular bone marrow composition in control and treated mice euthanized at week 24. Shown is the percentage of lineage marker-positive cells (Ter119+, CD3+, CD19+, and Gr-1+ cells). The differences between the 2 groups were not significant. BA, basophils; EO, eosinophils; Hb, hemoglobin; HCT, hematocrit; LY, lymphocytes; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; NE, neutrophils; RDW, red cell distribution width.
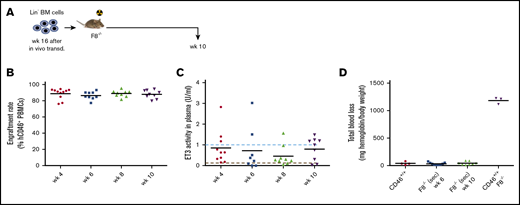
To demonstrate that we targeted primitive HSCs with our vector, we transplanted bone marrow Lin− cells harvested at week 24 after in vivo transduction into lethally irradiated secondary C57Bl/6 recipients and followed them for 16 weeks (supplemental Figure 5A). Engraftment rates were nearly 100% and remained stable (supplemental Figure 5B). Average plasma ET3 levels were stable and in the range of physiological levels (supplemental Figure 5C). Distribution of bone marrow lineage–positive cells of transplanted mice was similar to that of naive control mice (supplemental Figure 5D).
In summary, the outcome of the studies in the hCD46-transgenic model, specifically the finding that plasma ET3 levels were in the microgram/milliliter range, exemplified the ability to express a transgene product at high levels in erythroid cells and to release it into the bloodstream. Notably, because primitive HSCs were transduced, ET3 was expressed long term (24 weeks in in vivo transduced mice + 16 weeks in secondary recipients) without affecting erythropoiesis.
Ex vivo HSC gene therapy with HDAd-LCR-ET3/mgmt in a murine hemophilia A model
To demonstrate the practical relevance of expressing ET3 from erythroid cells, we attempted to correct the bleeding defect in hemophilia A mice. The model is based on 129S-F8tm1Kaz/J mice completely lacking (mouse) FVIII activity. For in vivo transduction with HDAd5/35++ vectors, we crossed 129S-F8tm1Kaz/J mice with hCD46tg mice (supplemental Figure 6A). The resulting hCD46+/−/F8−/− and hCD46+/+/F8−/− mice had no background FVIII plasma activity (supplemental Figure 6B) and were unable to stop bleeding after tail clipping (supplemental Figure 6C). hCD46/F8−/− mice were more fragile than the parental F8−/− strain and displayed spontaneous bleeding tendency before ET3 expression commenced. As outlined in the Introduction, ex vivo HSC gene therapy using lentivirus vectors showed preclinical promise.16,,-19 We therefore first tested in an ex vivo HSC transduction setting whether our HDAd5/35++ vector system could also achieve phenotypic correction when ET3 is expressed from erythroid cells. Ex vivo transduced bone marrow Lin− cells from hCD46+/−/F8−/− mice were transplanted into lethally irradiated F8−/− mice (Figure 5A). The engraftment rate was ∼90% in all mice at week 4 after transplantation (supplemental Figure 7). Transplanted animals received 4 rounds of in vivo selection and were euthanized at week 18 posttransplantation. Four out of 7 mice were lost around week 6 (before the onset of in vivo selection and substantial ET3 production) due to spontaneous bleeding after drug injection. The average plasma ET3 activity in the remaining 3 mice reached physiological levels at week 12 (Figure 5B). ET3 mRNA levels in total blood cells were comparably high for all 3 animals (Figure 5C). Also, in these animals, a complete phenotypic correction based on blood loss after tail clipping was observed (Figure 5D). Correction was also reflected in another functional clotting test that measures thrombin/antithrombin levels (Figure 5E).
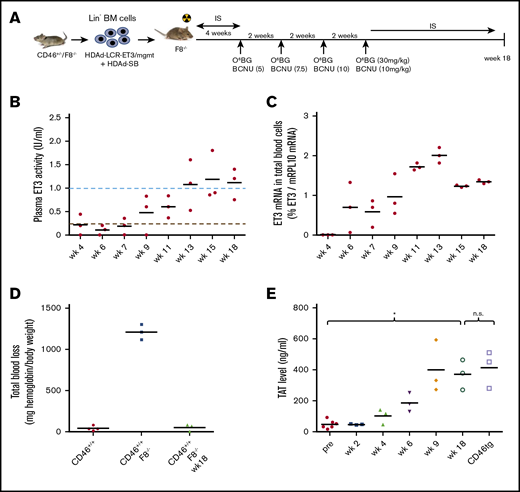
Phenotypic correction of hemophilia after ex vivo HSC transduction of hCD46+/−/F8−/−Lin−cells and transplantation into hemophilia A F8−/−mice. (A) Schematic of the experiment: bone marrow was harvested from hCD46+/−/F8−/− mice, and lineage-negative cells (Lin−) were isolated by MACS. Lin− cells were transduced with HDAd-LCR-ET3/mgmt and HDAd-SB alone at a total multiplicity of infection of 500 vp per cell. After 1 day in culture, 1 × 106 transduced cells per mouse were transplanted into lethally irradiated F8−/− mice. At week 4, O6BG/BCNU treatment was started and repeated 4 times every 2 weeks. With each cycle, the BCNU concentration was increased from 5 mg/kg, to 7.5 mg/kg, to 10 mg/kg (twice). At week 18, mice were euthanized. Before and after in vivo selection, mice received immunosuppressive drugs. BM, bone marrow. (B) ET3 activity measured in plasma by Coamatic FVIII assay. Activities of 100% and 5% are indicated by dotted lines. (C) ET3 mRNA levels in peripheral blood RBCs measured by qRT-PCR relative to mouse mRPL10 mRNA levels. (D) Bleeding after tail clipping. Blood was collected in saline over 45 minutes, and the hemoglobin amount was determined and expressed as milligrams normalized to body weight. (E) Concentrations of TAT complexes in plasma. Plasma from hCD46-transgenic mice served as control for physiological levels. Mice transplanted with ex vivo transduced HSCs were kept alive without side effects for 18 weeks. *P < .05. n.s., not significant.
Phenotypic correction of hemophilia after ex vivo HSC transduction of hCD46+/−/F8−/−Lin−cells and transplantation into hemophilia A F8−/−mice. (A) Schematic of the experiment: bone marrow was harvested from hCD46+/−/F8−/− mice, and lineage-negative cells (Lin−) were isolated by MACS. Lin− cells were transduced with HDAd-LCR-ET3/mgmt and HDAd-SB alone at a total multiplicity of infection of 500 vp per cell. After 1 day in culture, 1 × 106 transduced cells per mouse were transplanted into lethally irradiated F8−/− mice. At week 4, O6BG/BCNU treatment was started and repeated 4 times every 2 weeks. With each cycle, the BCNU concentration was increased from 5 mg/kg, to 7.5 mg/kg, to 10 mg/kg (twice). At week 18, mice were euthanized. Before and after in vivo selection, mice received immunosuppressive drugs. BM, bone marrow. (B) ET3 activity measured in plasma by Coamatic FVIII assay. Activities of 100% and 5% are indicated by dotted lines. (C) ET3 mRNA levels in peripheral blood RBCs measured by qRT-PCR relative to mouse mRPL10 mRNA levels. (D) Bleeding after tail clipping. Blood was collected in saline over 45 minutes, and the hemoglobin amount was determined and expressed as milligrams normalized to body weight. (E) Concentrations of TAT complexes in plasma. Plasma from hCD46-transgenic mice served as control for physiological levels. Mice transplanted with ex vivo transduced HSCs were kept alive without side effects for 18 weeks. *P < .05. n.s., not significant.
In vivo HSC gene therapy with HDAd-LCR-ET3/mgmt in a murine hemophilia A model
hCD46+/+/F8−/− hemophilia A mice were mobilized and IV injected with HDAd-LCR-ET3/mgmt (Figure 6A). Four cycles of in vivo selection with O6BG/BCNU were initiated 4 weeks later, and animals were followed for 16 weeks. If uncontrolled bleeding occurred during the injections up to week 6, recombinant ET3 protein was given IP (in 6 out of 10 mice). Compared with the in vivo HSC transduction study in hCD46tg mice (Figure 2), in hCD46+/+/F8−/− mice, plasma anti-ET3 antibody titers were ∼3 orders of magnitude (in mice that did not receive rET3) and ∼5 orders of magnitude (in mice that were injected with rET3) higher (Figure 6B). This prevented the measurement of ET3 plasma concentrations by ELISA. ET3 mRNA levels in total blood cells (∼0.6% ET3 of mRPL10mRNA) (Figure 6C) and bone marrow cells (∼40% ET3 of mRPL10mRNA) (Figure 6D) were comparable to those observed in the study with hCD46-transgenic mice in Figure 2. The immunoprecipitation/western blot assay (that disrupts ET3/antibody complexes) demonstrated the presence of ET3 protein in total blood cells (Figure 6E). In the 4 animals that did not receive rET3 injection early in the study, despite high-level plasma anti-ET3 antibodies, curative bleeding control was achieved (Figure 6E). This indicates that not all of the produced ET3 is bound to antibodies. No significant bleeding correction was observed in mice that had received multiple injection of rET3 protein, suggesting that the high antibody titers in these animals saturated all free plasma ET3 (data not shown). ET3 expression was predominantly in erythroid cells as mRNA analysis in bone marrow Ter119+ and Ter119- fractions suggested (Figure 6G). On average, ∼1.5 integrated vector copies per cell were found in bone marrow cells (Figure 6H). Bone marrow Lin− cells harvested at week 16 from in vivo transduced hCD46+/+/F8−/− mice were transplanted into irradiated (secondary) hemophilia A (F8−/−) mice (Figure 7A). Average engraftment rates in recipients were ∼90% (Figure 7B). The average ET3 plasma activity was ∼90% of physiological levels at week 10 (Figure 7C). In animals with declining ET3 activity, plasma anti-ET3 were detected at titers ∼102 at week 6, indicating that myeloablation by irradiation might not have been complete. Again, regardless of inhibitor antibody development, all mice displayed a fully corrected phenotype in the tail clipping assay (Figure 7D).

Phenotypic correction of hemophilia after in vivo HSC transduction of hCD46+/+/F8−/−mice. (A) Schematic of the experiment. hCD46+/+/F8−/− mice were mobilized and in vivo transduced as described in Figure 2. Mice received 4 rounds of O6BG/BCNU treatment and were euthanized at week 16 after in vivo transduction. (B) Plasma anti-ET3 antibody titers in representative mice of the 4 experimental settings tested. Black curves, representative antibody titers in hCD46-transgenic mice after in vivo HSC transduction (see Figure 2). Green curve, representative antibody titers in hCD46+/+/F8−/− mice after in vivo transduction; red curves, antibody titers in hCD46+/+/F8−/− mice after in vivo transduction, whereby mice received recombinant ET3 protein injections to control bleeding during blood sampling; blue curves, representative antibody titers in hCD46+/+/F8−/− mice after ex vivo transduction. (C) ET3 mRNA in peripheral blood RBCs measured by qRT-PCR relative to mouse mRPL10 mRNA levels. (D) ET3 mRNA levels in bone marrow mononuclear cells at week 16 measured by qRT-PCR relative to mouse mRPL10 mRNA levels. (E) Immunoprecipitation/western blot with lysates from total blood cells (ie, mostly erythrocytes). The specific ET3 band appears ∼168 kDa. (F) Bleeding after tail clipping. Blood was collected in saline over 45 minutes, and the hemoglobin amount was determined and expressed as milligrams normalized to body weight. (G) ET3 mRNA in bone marrow erythroid Ter119+ and nonerythroid Ter119− cells at week 16 relative to mouse mRPL10 mRNA levels. (H) VCN per cell in bone marrow MNCs at week 16 after in vivo HSC transduction. In vivo HSC transduced mice were kept alive without side effects for 16 weeks.
Phenotypic correction of hemophilia after in vivo HSC transduction of hCD46+/+/F8−/−mice. (A) Schematic of the experiment. hCD46+/+/F8−/− mice were mobilized and in vivo transduced as described in Figure 2. Mice received 4 rounds of O6BG/BCNU treatment and were euthanized at week 16 after in vivo transduction. (B) Plasma anti-ET3 antibody titers in representative mice of the 4 experimental settings tested. Black curves, representative antibody titers in hCD46-transgenic mice after in vivo HSC transduction (see Figure 2). Green curve, representative antibody titers in hCD46+/+/F8−/− mice after in vivo transduction; red curves, antibody titers in hCD46+/+/F8−/− mice after in vivo transduction, whereby mice received recombinant ET3 protein injections to control bleeding during blood sampling; blue curves, representative antibody titers in hCD46+/+/F8−/− mice after ex vivo transduction. (C) ET3 mRNA in peripheral blood RBCs measured by qRT-PCR relative to mouse mRPL10 mRNA levels. (D) ET3 mRNA levels in bone marrow mononuclear cells at week 16 measured by qRT-PCR relative to mouse mRPL10 mRNA levels. (E) Immunoprecipitation/western blot with lysates from total blood cells (ie, mostly erythrocytes). The specific ET3 band appears ∼168 kDa. (F) Bleeding after tail clipping. Blood was collected in saline over 45 minutes, and the hemoglobin amount was determined and expressed as milligrams normalized to body weight. (G) ET3 mRNA in bone marrow erythroid Ter119+ and nonerythroid Ter119− cells at week 16 relative to mouse mRPL10 mRNA levels. (H) VCN per cell in bone marrow MNCs at week 16 after in vivo HSC transduction. In vivo HSC transduced mice were kept alive without side effects for 16 weeks.

Analysis of secondary F8−/−recipients of Lin−cells from in vivo transduced hCD46+/+F8−/−mice. (A) Bone marrow Lin− cells pooled from 4 CD46+/+/F8−/− mice at week 16 after in vivo transduction (Figure 6) were transplanted into 1050 Rad irradiated F8−/− hemophilia A mice. (B) Engraftment based on the percentage of hCD46-positive peripheral blood mononuclear cells (PBMCs). (C) ET3 activity measured in plasma by Coamatic FVIII assay. Activities of 100% and 5% are indicated by dotted lines. (D) Bleeding after tail clipping analyzed at week 10 after transplantation. For comparison, “healthy” CD46+/+ and hemophilia A CD46+/+/F8−/− mice are shown. Secondary recipients were kept alive without side effects for 10 weeks.
Analysis of secondary F8−/−recipients of Lin−cells from in vivo transduced hCD46+/+F8−/−mice. (A) Bone marrow Lin− cells pooled from 4 CD46+/+/F8−/− mice at week 16 after in vivo transduction (Figure 6) were transplanted into 1050 Rad irradiated F8−/− hemophilia A mice. (B) Engraftment based on the percentage of hCD46-positive peripheral blood mononuclear cells (PBMCs). (C) ET3 activity measured in plasma by Coamatic FVIII assay. Activities of 100% and 5% are indicated by dotted lines. (D) Bleeding after tail clipping analyzed at week 10 after transplantation. For comparison, “healthy” CD46+/+ and hemophilia A CD46+/+/F8−/− mice are shown. Secondary recipients were kept alive without side effects for 10 weeks.
In summary, the studies in hemophilia A mice corroborate findings made in the hCD46-transgenic model. The ET3 level was high enough to saturate anti-ET3 inhibitor antibodies (up to titers of ∼5 × 105) and provide sufficient noninhibited ET3 systemically and/or locally (in blood clots) so that a phenotypic correction of the bleeding disorder was achieved.
Discussion
The goal of this study was to capitalize on the enormous capacity of the hematopoietic system to generate erythroid cells and explore the possibility of producing therapeutic proteins from HSC-derived erythroid cells. Instrumental for achieving this goal was a novel in vivo HSC transduction/selection approach and the use of β-globin regulatory elements (ie, a mini-LCR and the 3′UTR) for transgene expression. Furthermore, we speculated that erythrocytes trapped in blood clots would locally release ET3, which would then contribute to the control of bleeding in the presence of inhibitor antibodies.
Because of their abundance and systemic distribution, erythrocytes have been used for decades as a controlled drug delivery system.6,27 Several ex vivo drug-loading strategies have been pursued, including (1) internal loading of drugs by penetration through the RBC membrane, for example, by hypotonic loading, cell-penetrating cargo, or fusion with liposomes; (2) surface loading by chemical coupling and binding of RBC-targeted affinity ligands; and (3) differentiation of engineered, enucleated RBCs derived from stem cells (peripheral blood CD34+ cells, iPSCs, etc).27 The approach to express a nonerythroid protein (factor IX) from HSC-derived erythroid cells was pioneered by the Sadelain group.28 Their strategy involved the ex vivo transduction of HSCs with a lentivector and subsequent transplantation into irradiated recipients. We used a simplified HSC transduction approach that is also potentially less expensive and applicable to larger patient populations. Because of the 30-kb insert capacity of our HDAd5/35++ gene transfer vector platform, we were able to incorporate an in vivo selection cassette (mgmtP140K) as well as regulatory elements capable of mediating high-level transgene expression in erythroid cells. An indication of the high efficacy of our in vivo HSC transduction/selection approach was the finding that 100% of CFUs in the bone marrow contained integrated vector DNA. Based on mRNA levels, we demonstrated that the protein is produced predominantly in erythroid cells in the bone marrow and stored in peripheral blood RBCs at high concentrations with up 90% to 100% GFP+ RBCs in some animals.
In vivo HSC transduction/selection in hCD46-transgenic mice using an LCR-ET3–containing HDAd5/35++ resulted in supraphysiological plasma ET3 levels. Despite these high ET3 levels, no detrimental effects on erythropoiesis were observed, most likely due to efficient secretion/release of ET3 from erythroid cells. Furthermore, hemoglobin analyses in RBCs did not reveal abnormalities, suggesting that the additional new mini-LCR copies (∼1.5-2 per cell) did not saturate the transcriptional machinery in erythroid cells. We hypothesize that ET3 detected in peripheral blood is the product of active secretion from erythroid cells in the bone marrow as well as the release from senescent erythrocytes. The latter involves the loss of membrane in the form of microvesicles resulting in an increased cellular density and fragility. We followed groups of mice for 24 weeks without observing a significant decrease in RBC numbers or shape arguing against the possibility that added ET3 production accelerated senescence. The question whether ET3 released from senescent erythrocytes has undergone structural changes and reduction of activity remains to be studied.
In the hCD46-transgenic model, we also observed signs of induction of tolerance against ET3 (eg, absence or disappearance of plasma antibodies) in the majority of mice. We speculate that this is triggered by low-level ET3 expression in tolerogenic thymocytes. Similar high ET3 expression levels (based on mRNA) were observed after in vivo HSC transduction of hCD46+/+/F8−/− hemophilia A mice. Unlike hCD46-transgenic mice, all hemophilia mice developed a strong humoral immune response against the neoantigen ET3 upon in vivo HSC gene therapy. Except in hCD46+/+/F8−/− mice that received additional recombinant ET3 protein injections, the high plasma anti-ET3 titers did not compromise the therapeutic effect of our approach (ie, the phenotyping correction of bleeding). We speculate that not all ET3 is in complex with inhibitory antibodies or that ET3 is released locally from erythrocytes at the site of injury or as a part of the blood clot triggered efficient coagulation. The strong humoral anti-ET3 immune response observed in hemophilia A mice was not found in studies after nonmyeloablative transplantation of ex vivo transduced HSCs.17,-19 It is possible that the relatively stringent erythroid specificity of ET3 expression from our vector was suboptimal for tolerance induction in hemophilia A mice. The lower degree of humoral immune response in hCD46-transgenic mice could, in part, be explained by the fact that mouse FVIII and ET3 are 80% homologous and therefore, most likely, share antigen epitopes.
SB100x transposase mediates random transgene integration.4,24 Although this integration pattern is theoretically safer than that of lenti- and rAAV vectors, which predominantly integrate into active genes/promoters,29,30 it still raises concerns with regards to transgene silencing, undesired effects on neighboring genes, and genomic rearrangements. Approaches capable of directing transgene integration to a preselected “safe harbor” locus that involve the ex vivo transduction of HSCs with endonuclease-encoding mRNA and rAAV6 vectors 31,-33 have been successfully tested. We have recently used our in vivo HSC transduction approach for targeted transgene integration into the AAVS1 locus in mice .34
Our study is the first demonstration that in vivo HSC gene therapy with a therapeutic plasma protein results in phenotypic correction in a mouse disease model. In addition to FVIII, the application of our approach for other plasma proteins can be envisioned provided that they do not exert detrimental effects on erythropoiesis: (1) other coagulation factors, specifically FXI, FVII,35 von Willebrand factor,36 but also rare clotting factors (ie, factors I, II, V, X, XI, or XIII); (2) enzymes that are currently used in enzyme replacement therapies for lysosomal storage diseases (taking advantage of the cross-correction mechanism),37 like Pompe disease (acid α-glucosidase), Gaucher disease (glucocerebrosidase), Fabry disease (α-galactosidase A), and Mucopolysaccharidosis type I (α-L-Iduronidase); (3) immunodeficiencies (eg, SCID-ADA38 [adenosine deaminase]); (4) cardiovascular diseases (eg, familial apolipoprotein E deficiency and atherosclerosis [ApoE])39 ; (5) viral infections by expression of viral decoy receptors (eg, for HIV, soluble CD4),40 or broadly neutralizing antibodies (bNAbs) for HIV,41 chronic HCV,42 or HBV43 infections; and (6) cancer (eg, controlled expression of monoclonal antibodies, eg, trastuzumab44 ) or checkpoint inhibitors (eg, aPDL145 ).
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Chandana Kulkarni and Lexus Pina for help in the animal studies.
This study was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01HL141781 and R01HL128288, and a grant from the Marsha Rivkin Foundation (A.L.).
Authorship
Contribution: A.L. provided the conceptual framework for the study, designed the experiments, and wrote the manuscript; H.W., Z.L., C.L., and S.G. performed the experiments; C.B.D. provided the ET3 construct and critical help; and T.P. and C.B.D. provided critical comments on the manuscript;.
Conflict-of-interest disclosure: C.B.D. is a cofounder of Expression Therapeutics and owns equity in the company. Expression Therapeutics owns the intellectual property associated with ET3. The terms of this arrangement have been reviewed and approved by Emory University in accordance with its conflict of interest policies. The remaining authors declare no competing financial interests.
Correspondence: André Lieber, University of Washington, 1705 NE Pacific St, Seattle, WA 98195; e-mail: lieber00@uw.edu.