Key Points
RA synergizes with the N-glycosylation inhibitor tunicamycin and ATO to induce AML cell death via generation of ER and oxidative stress.

Abstract
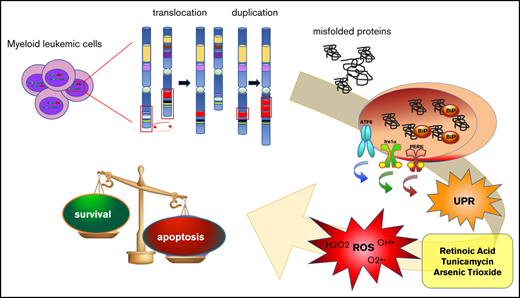
Acute myeloid leukemia (AML) is often characterized by the expression of fusion or mutant proteins that cause impaired differentiation and enhanced proliferation and survival. The presence of mutant proteins prone to misfolding can render the cells sensitive to endoplasmic reticulum (ER) stress and oxidative stress that could otherwise be overcome. Here, we show that the triple combination of the differentiating agent retinoic acid (RA), the ER stress–inducing drug tunicamycin (Tm), and arsenic trioxide (ATO), able to generate oxidative stress, leads to the death of AML cell lines expressing fusion proteins involving the gene MLL and the internal tandem duplication (ITD) in the FLT3 tyrosine kinase receptor. Importantly, the combination of RA, Tm, and ATO decreased the colony-forming capacity of primary leukemic blasts bearing the FLT-ITD mutation without affecting healthy hematopoietic progenitor cells. We demonstrate in cell lines that combination of these drugs generates ER and oxidative stresses and impairs maturation and causes accumulation of FLT3 protein in the ER. Our data provide a proof of concept that low amounts of drugs that generate ER and oxidative stresses combined with RA could be an effective targeted therapy to hit AML cells characterized by MLL fusion proteins and FLT3-ITD mutation.
Introduction
Present therapies for acute myeloid leukemia (AML) provide a rate of cure of 40% to 50%; therefore, novel approaches are needed.1 Endoplasmic reticulum (ER) stress triggers the unfolded protein response (UPR), which plays an essential role in maintaining protein homeostasis (proteostasis). The concept of perturbing proteostasis to promote cancerous cell death has been extensively described in multiple myeloma.2 We demonstrated that the ER stress–inducing drug tunicamycin (Tm) led to acute promyelocytic leukemia cell death in synergy with the differentiation agent retinoic acid (RA) and arsenic trioxide (ATO), which generates oxidative stress,3 at low doses of each drug, which had little or no effect when used alone. Furthermore, the acute promyelocytic leukemia oncogenic fusion protein PML-RARα formed intracellular protein aggregates upon treatment with RA and Tm, further exacerbating stress of the secretory protein folding compartment. Thus, mutant proteins, characterizing a variety of AMLs, could provide the basis of high sensitivity to drug-induced disruption of proteostasis, because they are often a source of proteostasis imbalance. For example, the mixed lineage leukemia (MLL) protein is a histone methyltransferase found with >60 fusion partners generating various types of leukemia.4 In particular, the MLL-AF6 fusion protein sequesters AF6 into the nucleus from its normal cytosolic localization.5 The internal tandem duplication in FMS-like tyrosine kinase 3 (FLT3-ITD) is one of the most common mutations in AML, found in ∼30% of patients, and associated with a poor outcome.6 FLT3-ITD is particularly interesting in this context, because it is a misfolded protein mostly retained in the ER.7 In this work, we set out to explore the sensitivity of AML cells expressing different oncogenic proteins to the combination of low doses of RA, Tm, and ATO (RTA).
Methods
Cell culture
ML-2, MV-4-11, MOLM13, THP1, SKNO-1, HL60, HEL, U937, OCI-AML-2, and OCI-AML-3 cell lines were treated with 10 nM RA, 50 ng/mL of Tm, 500 nM ATO, 20 mM N-acetylcysteine (NAC), and 2.5 mM 4-PBA alone or in combination, as indicated in figures for each experiment. Primary cells isolated from bone marrow were cultured in Methocult 4035 medium with or without 10 nM RA, 50 ng/mL of Tm, and 500 nM ATO alone or in combination.
Cell death and differentiation
Cell death was evaluated by propidium iodide exclusion assay. Cell differentiation of AML primary blasts and healthy bone marrow cells, grown in Methocult medium for 8 days, was assessed by morphological analysis of cytospin preparations stained with Wright-Giemsa.
Immunofluorescence
Confocal microscopy was performed on cytospin preparations stained with primary anti-FLT3 and anti-BiP antibodies followed by anti-rabbit Alexa Fluor-488 or anti-mouse Alexa Fluor-555. DNA was counterstained with TOPRO-3. The images were acquired with the Leica laser scanning microscope TCS SP2 using the 40× objective.
Quantitative real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction was analyzed by the ΔΔCt method using histone 3 or glyceraldehyde-3-phosphate dehydrogenase as endogenous control for standardization.
Western blot
Total protein extract (20-40 μg) was separated by SDS–PAGE, in reducing or nonreducing conditions.
Detailed materials and methods are available in the supplemental Material.
Results and discussion
Among a panel of AML cells carrying different oncogenic mutant proteins, the ML-2, MOLM-13, and MV-4-11 cells, expressing the fusion proteins MLL-AF6, MLL-AF9 plus FLT3-ITD, and MLL-AF4 plus FLT3-ITD, respectively, were the most sensitive to the RTA combination; sensitivity was more significant in the ML-2 and MV-4-11 cells (Figure 1A; supplemental Figure 1A; supplemental Table 1). It is worth noticing that the pair comprising THP1 and ML-2, expressing MLL fusion proteins, and the pair comprising MOLM-13 and MV-4-11, expressing MLL fusion proteins and FLT3-ITD, showed similar patterns of response to the different treatments. ML-2 and MV-4-11 cells exhibited a diverse sensitivity to RA that was not surprising, considering the wide range of responses to RA of different types of AML described in literature.8 When we treated primary leukemic cells with the same drugs, we found that the RTA combination significantly impaired the clonogenic capacity of primary cells harvested from the bone marrow of FLT3-ITD+ AML patients, and morphological analysis revealed that a high percentage of RTA-treated cells presented nuclear vacuoles, suggesting cell death (Figure 1B upper panel; Figure 1C). However, the same combination had no significant effect on leukemic blasts negative for the FLT3-ITD mutation (Figure 1B middle panel). Although these analyses must be extended to a higher number of samples, they suggest that AML primary FLT3-ITD+ cells are sensitive to the RTA combination, independently of the presence of additional mutations (supplemental Table 2). Importantly, the effects of RTA were specific to leukemic cells, because the same combination did not alter the clonogenic capacity of hematopoietic progenitor cells isolated from the bone marrow of healthy donors (Figure 1B lower panel; supplemental Figure 1).
![Combination of ER and oxidative stress with RA induces AML cell death and intracellular accumulation of immature FLT3. (A) ML-2 and MV-4-11 cells, treated for 72 hours with 10 nM RA (R), 50 ng/mL of Tm (T), and 500 nM ATO (A) alone or in combination, were analyzed for propidium iodide (PI) uptake to evaluate cell death (n = 16 ± standard error of the mean [SEM]). One-way ANOVA vs C (control, vehicle-treated cells): **P < .005, ***P < .001, ****P < .0001. Student t test ####P < .0001. Synergy of RA, Tm, and ATO was confirmed by the Chou-Talalay method21 (combination index in MV-4-11 cells, 0.34). (B) Colony-forming unit assay for AML blasts isolated from the bone marrow (BM) of 6 patients who were FLT3-ITD+ and 3 who were FLT-ITD− and for mononucleated cells isolated from 4 healthy donors. The cells were treated in semisolid medium with 10 nM RA, 50 ng/mL of Tm, and 500 nM ATO alone or in combination as indicated. After 8 days, the colony size (number of cells per colony) was evaluated by microscopy. The graphs report the ratio of the average number of cells forming the colonies of each treated sample over its control. Student t test ***P < .001. (C) Morphological analysis of AML1 cells isolated from 8-day colonies obtained in panel B. Black arrows indicate cell vacuoles. The graph at the bottom reports the average ratio of the number of vacuolated cells over those nonvacuolated (n = 3 ± SEM). One-way ANOVA vs C: *P < .05, ***P < .001. Student t test of TA vs RTA: †P< .05. (D) Western blot of ML-2 and MV-4-11 protein extracts from cells treated with vehicle, C, or with the combination RTA, as in panel A, for 48 hours to detect FLT3. Control ML-2 cells had no detectable levels of FLT3; FLT3 was instead accumulated in immature, not fully glycosylated forms upon treatment with RTA. MV-4-11 cells carry a homozygous FLT3-ITD mutation that is mostly retained intracellularly in immature forms (C). Treatment of cells with RTA further impaired FLT3-ITD glycosylation, as demonstrated by the disappearance of the fully glycosylated (mature) form and by the increase of the less glycosylated form. (E) Confocal analysis of the distribution of FLT3 and BiP proteins in ML-2 and MV-4-11 cells in control and RTA-treated cells. According to the observations obtained by western blot analysis in panel A, FLT3 is found on the plasma membrane of ML-2 control cells and accumulates in the ER upon treatment with RTA. BiP is the main ER chaperone, and colocalization of FLT3 with BiP demonstrates misfolding and retention in the ER (yellow spots in inset 1). FLT3-ITD is already retained intracellularly in control MV-4-11 cells, and treatment with RTA increases the amount of FLT3-ITD in the ER (inset 2). ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/24/10.1182_bloodadvances.2019000540/3/m_advancesadv2019000540f1.png?Expires=1763535269&Signature=5DxnrK1-Wg~9got3XuGKKT6tzfGsMweMTTtx0nJWiRwcoMouWhaWUb-JQiVxETItyPfNbBY6QE~SjBoSazbMJZHNnJWsEDms7EXUyMO3QtbxbvtNZBwC-zfIlkrpLUOFjp-vsS0tyD39XSP4YkafbPWYS7X23P3Paudg4xF~fu4EbV8D7w3mgWxpE4Rua7~Q1dlZCPDRqceIu8diODJq3hz6vOYbIXxqC~2CkSMk589EQXRLxKqX5CRFLZQ1m1O2du8FS1IKwe3nv0qDkYRFBDQbyym95eAuJFDBdWwBuAcnZmYAxE5eUfEcLdLpjBsHKMOxDNFpCB3sKE9BFKYZ~A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Combination of ER and oxidative stress with RA induces AML cell death and intracellular accumulation of immature FLT3. (A) ML-2 and MV-4-11 cells, treated for 72 hours with 10 nM RA (R), 50 ng/mL of Tm (T), and 500 nM ATO (A) alone or in combination, were analyzed for propidium iodide (PI) uptake to evaluate cell death (n = 16 ± standard error of the mean [SEM]). One-way ANOVA vs C (control, vehicle-treated cells): **P < .005, ***P < .001, ****P < .0001. Student t test ####P < .0001. Synergy of RA, Tm, and ATO was confirmed by the Chou-Talalay method21 (combination index in MV-4-11 cells, 0.34). (B) Colony-forming unit assay for AML blasts isolated from the bone marrow (BM) of 6 patients who were FLT3-ITD+ and 3 who were FLT-ITD− and for mononucleated cells isolated from 4 healthy donors. The cells were treated in semisolid medium with 10 nM RA, 50 ng/mL of Tm, and 500 nM ATO alone or in combination as indicated. After 8 days, the colony size (number of cells per colony) was evaluated by microscopy. The graphs report the ratio of the average number of cells forming the colonies of each treated sample over its control. Student t test ***P < .001. (C) Morphological analysis of AML1 cells isolated from 8-day colonies obtained in panel B. Black arrows indicate cell vacuoles. The graph at the bottom reports the average ratio of the number of vacuolated cells over those nonvacuolated (n = 3 ± SEM). One-way ANOVA vs C: *P < .05, ***P < .001. Student t test of TA vs RTA: †P< .05. (D) Western blot of ML-2 and MV-4-11 protein extracts from cells treated with vehicle, C, or with the combination RTA, as in panel A, for 48 hours to detect FLT3. Control ML-2 cells had no detectable levels of FLT3; FLT3 was instead accumulated in immature, not fully glycosylated forms upon treatment with RTA. MV-4-11 cells carry a homozygous FLT3-ITD mutation that is mostly retained intracellularly in immature forms (C). Treatment of cells with RTA further impaired FLT3-ITD glycosylation, as demonstrated by the disappearance of the fully glycosylated (mature) form and by the increase of the less glycosylated form. (E) Confocal analysis of the distribution of FLT3 and BiP proteins in ML-2 and MV-4-11 cells in control and RTA-treated cells. According to the observations obtained by western blot analysis in panel A, FLT3 is found on the plasma membrane of ML-2 control cells and accumulates in the ER upon treatment with RTA. BiP is the main ER chaperone, and colocalization of FLT3 with BiP demonstrates misfolding and retention in the ER (yellow spots in inset 1). FLT3-ITD is already retained intracellularly in control MV-4-11 cells, and treatment with RTA increases the amount of FLT3-ITD in the ER (inset 2). ns, not significant.
Combination of ER and oxidative stress with RA induces AML cell death and intracellular accumulation of immature FLT3. (A) ML-2 and MV-4-11 cells, treated for 72 hours with 10 nM RA (R), 50 ng/mL of Tm (T), and 500 nM ATO (A) alone or in combination, were analyzed for propidium iodide (PI) uptake to evaluate cell death (n = 16 ± standard error of the mean [SEM]). One-way ANOVA vs C (control, vehicle-treated cells): **P < .005, ***P < .001, ****P < .0001. Student t test ####P < .0001. Synergy of RA, Tm, and ATO was confirmed by the Chou-Talalay method21 (combination index in MV-4-11 cells, 0.34). (B) Colony-forming unit assay for AML blasts isolated from the bone marrow (BM) of 6 patients who were FLT3-ITD+ and 3 who were FLT-ITD− and for mononucleated cells isolated from 4 healthy donors. The cells were treated in semisolid medium with 10 nM RA, 50 ng/mL of Tm, and 500 nM ATO alone or in combination as indicated. After 8 days, the colony size (number of cells per colony) was evaluated by microscopy. The graphs report the ratio of the average number of cells forming the colonies of each treated sample over its control. Student t test ***P < .001. (C) Morphological analysis of AML1 cells isolated from 8-day colonies obtained in panel B. Black arrows indicate cell vacuoles. The graph at the bottom reports the average ratio of the number of vacuolated cells over those nonvacuolated (n = 3 ± SEM). One-way ANOVA vs C: *P < .05, ***P < .001. Student t test of TA vs RTA: †P< .05. (D) Western blot of ML-2 and MV-4-11 protein extracts from cells treated with vehicle, C, or with the combination RTA, as in panel A, for 48 hours to detect FLT3. Control ML-2 cells had no detectable levels of FLT3; FLT3 was instead accumulated in immature, not fully glycosylated forms upon treatment with RTA. MV-4-11 cells carry a homozygous FLT3-ITD mutation that is mostly retained intracellularly in immature forms (C). Treatment of cells with RTA further impaired FLT3-ITD glycosylation, as demonstrated by the disappearance of the fully glycosylated (mature) form and by the increase of the less glycosylated form. (E) Confocal analysis of the distribution of FLT3 and BiP proteins in ML-2 and MV-4-11 cells in control and RTA-treated cells. According to the observations obtained by western blot analysis in panel A, FLT3 is found on the plasma membrane of ML-2 control cells and accumulates in the ER upon treatment with RTA. BiP is the main ER chaperone, and colocalization of FLT3 with BiP demonstrates misfolding and retention in the ER (yellow spots in inset 1). FLT3-ITD is already retained intracellularly in control MV-4-11 cells, and treatment with RTA increases the amount of FLT3-ITD in the ER (inset 2). ns, not significant.
We then investigated the stress responses activated by RTA. It is established that the ITD of FLT3 impairs its full glycosylation and folding, causing its retention in the ER.7 Accordingly, we observed that FLT3-ITD is partially retained in the ER in an immature form and that treatment with RTA caused further hindrance of protein maturation and retention in the ER of FLT3-ITD in MV-4-11 cells, as well as of wild-type FLT3 in ML-2 cells (Figure 1D-E). Instead, Tm alone increased the ratio between underglycosylated and mature FLT3-ITD only slightly (supplemental Figure 2), suggesting that only the RTA combination appreciably affected the folding efficiency of the ER. Nonetheless, Tm activated the UPR in ML-2 and MV-4-11 cells either alone or in combination with RA and/or ATO (Figure 2 A-D). Treatment with ATO alone triggered the oxidative stress response, which reached much higher levels upon administration of ATO in combination with RA and Tm, as shown by increased expression of the gene HMOX-1, a main player in this response (Figure 2A). ER stress and oxidative stress are tightly linked, even though the mechanisms of interaction between these stresses are still unknown.9 Indeed, generation of reactive oxygen species and consequent oxidative stress is one of the mechanisms triggered by the unfolded protein response to induce apoptosis in the presence of overwhelming ER stress10 ; furthermore, altered redox homeostasis induces ER stress.11 Our data suggest that the ER stress response triggered by Tm alone and the oxidative stress response triggered by ATO alone reestablish homeostasis, resulting in cell survival, whereas the combination of Tm with ATO and RA leads to overcoming levels of oxidative stress and consequent cell death. Indeed, the antioxidant agent NAC rescued the deleterious effect of the combination of TA and RTA on cell viability of ML-2 cells (Figure 2E) and reduced oxidative and ER stresses (Figure 2F; supplemental Figure 3). The chemical chaperone sodium-4-phenylbutyrate, which favors protein folding,12 produced similar, although less marked, effects than NAC (Figure 2E-F). The effects of NAC and sodium-4-phenylbutyrate on MV4-11 cells, treated with RTA, were comparable to those observed on ML-2 cells, but much milder (supplemental Figure 4), likely because of the major response to RA of these cells. Indeed, the molecular mechanisms underlying the effects of RA on the ER and oxidative stress responses constitute an open question, which will be important to investigate. Aside from the differences between the 2 cell lines, our data indicate that treatment of ML-2 and MV-4-11 cells with RTA activated the UPR and the oxidative stress response, leading to cell death.
![Treatment of ML-2 and MV-4-11 cells with the RTA combination triggers the UPR and the oxidative stress response. (A) The RNA of ML-2 and MV-4-11 cells, treated as described in Figure 1A for 24 or 72 hours, was analyzed by quantitative real-time polymerase chain reaction for the expression of UPR target genes (the transcription factor CHOP, the ER chaperone BiP, and the spliced, active form of the transcription factor XBP1 [sXBP1]) and for the expression of HMOX, a gene with a main role in the oxidative stress response. sXBP1 induces the expression of a plethora of genes that increase the ER folding capacity, whereas CHOP is involved in the proapoptotic branch of the UPR (ML-2, n = 5 ± SEM; MV-4-11, n = 3 ± SEM; ML-2 and MV-4-11 HMOX, n = 2 ± SEM). Student t test vs C: *P < .05, **P < .005, ***P < .001. Student t test vs RA: †P < .05, ††P < .005, †††P < .001. (B) Western blot of ML-2 and MV-4-11 protein extracts, from cells treated as in panel A for 24 or 72 hours, to detect CHOP and BiP proteins. The graphs below each western blot report densitometric quantification (n = 2 ± SEM). Student t test *P < .05. (C) Confocal analysis of the expression of the ER chaperone BiP in ML-2 and MV-4-11 cells vehicle or RTA treated. (D) Western blot of protein extracts from ML-2 and MV-4-11 cells vehicle or RTA treated for 72 hours to detect the complexes among the chaperone BiP and its client misfolded proteins. The 72-KDa BiP monomer is indicated by the arrow. Unbalance of ER proteostasis upon treatment with RTA determined accumulation of misfolded proteins bound by BiP, indicated as BiP complexes. (E) ML-2 cells, treated for 72 hours as in Figure 1A in the presence or not of 20 mM NAC or of 2.5 mM sodium-4-phenylbutyrate (PBA), were analyzed for propidium iodide (PI) uptake to evaluate cell death (NAC, n = 4 ± SEM; PBA, n = 3 ± SEM). Student t test *P < .05, ****P < .0001. (F) Western blot of protein extracts from ML-2 cells, treated as in panel A, to detect the BiP misfolded protein complexes. NAC relieved oxidative stress induced by RTA and rescued the functionality of the ER, as indicated by the reduction of BiP protein level and by the loss of BiP complexes. A similar effect, although in minor measure, was achieved by PBA.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/24/10.1182_bloodadvances.2019000540/3/m_advancesadv2019000540f2.png?Expires=1763535269&Signature=YsbIHTZlyhzB-aFMS2uBL5~3FNKFFvob7cd3LOYbdgyS4WoNApKsPPpyIuWHfhA8EqOaLqedJGzt6LvqEeCpVFuD5XRi5HglGb3pQhLqr~Giy39hGnjmyp4KfEUqU02V2UbiM48tNi95b4vf1B-JUWDrRPYQf46NKZeLttoE79KIyFzHfmpLQ~uPeHlWx6l8TZ5doY3iGTqEAx8DC4q1OUTfIvgQ8pcek5unD-70QQdmKdsueZIJxUyh0xsZHnjYo7AgMJ-gB43FZOibSwjEX3IPrU5vbZOo~xqWL9dwngQuqTCVHsNynLhpXQYuSDfqYBcdvqZvLjMUvcPnkoAFPg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Treatment of ML-2 and MV-4-11 cells with the RTA combination triggers the UPR and the oxidative stress response. (A) The RNA of ML-2 and MV-4-11 cells, treated as described in Figure 1A for 24 or 72 hours, was analyzed by quantitative real-time polymerase chain reaction for the expression of UPR target genes (the transcription factor CHOP, the ER chaperone BiP, and the spliced, active form of the transcription factor XBP1 [sXBP1]) and for the expression of HMOX, a gene with a main role in the oxidative stress response. sXBP1 induces the expression of a plethora of genes that increase the ER folding capacity, whereas CHOP is involved in the proapoptotic branch of the UPR (ML-2, n = 5 ± SEM; MV-4-11, n = 3 ± SEM; ML-2 and MV-4-11 HMOX, n = 2 ± SEM). Student t test vs C: *P < .05, **P < .005, ***P < .001. Student t test vs RA: †P < .05, ††P < .005, †††P < .001. (B) Western blot of ML-2 and MV-4-11 protein extracts, from cells treated as in panel A for 24 or 72 hours, to detect CHOP and BiP proteins. The graphs below each western blot report densitometric quantification (n = 2 ± SEM). Student t test *P < .05. (C) Confocal analysis of the expression of the ER chaperone BiP in ML-2 and MV-4-11 cells vehicle or RTA treated. (D) Western blot of protein extracts from ML-2 and MV-4-11 cells vehicle or RTA treated for 72 hours to detect the complexes among the chaperone BiP and its client misfolded proteins. The 72-KDa BiP monomer is indicated by the arrow. Unbalance of ER proteostasis upon treatment with RTA determined accumulation of misfolded proteins bound by BiP, indicated as BiP complexes. (E) ML-2 cells, treated for 72 hours as in Figure 1A in the presence or not of 20 mM NAC or of 2.5 mM sodium-4-phenylbutyrate (PBA), were analyzed for propidium iodide (PI) uptake to evaluate cell death (NAC, n = 4 ± SEM; PBA, n = 3 ± SEM). Student t test *P < .05, ****P < .0001. (F) Western blot of protein extracts from ML-2 cells, treated as in panel A, to detect the BiP misfolded protein complexes. NAC relieved oxidative stress induced by RTA and rescued the functionality of the ER, as indicated by the reduction of BiP protein level and by the loss of BiP complexes. A similar effect, although in minor measure, was achieved by PBA.
Treatment of ML-2 and MV-4-11 cells with the RTA combination triggers the UPR and the oxidative stress response. (A) The RNA of ML-2 and MV-4-11 cells, treated as described in Figure 1A for 24 or 72 hours, was analyzed by quantitative real-time polymerase chain reaction for the expression of UPR target genes (the transcription factor CHOP, the ER chaperone BiP, and the spliced, active form of the transcription factor XBP1 [sXBP1]) and for the expression of HMOX, a gene with a main role in the oxidative stress response. sXBP1 induces the expression of a plethora of genes that increase the ER folding capacity, whereas CHOP is involved in the proapoptotic branch of the UPR (ML-2, n = 5 ± SEM; MV-4-11, n = 3 ± SEM; ML-2 and MV-4-11 HMOX, n = 2 ± SEM). Student t test vs C: *P < .05, **P < .005, ***P < .001. Student t test vs RA: †P < .05, ††P < .005, †††P < .001. (B) Western blot of ML-2 and MV-4-11 protein extracts, from cells treated as in panel A for 24 or 72 hours, to detect CHOP and BiP proteins. The graphs below each western blot report densitometric quantification (n = 2 ± SEM). Student t test *P < .05. (C) Confocal analysis of the expression of the ER chaperone BiP in ML-2 and MV-4-11 cells vehicle or RTA treated. (D) Western blot of protein extracts from ML-2 and MV-4-11 cells vehicle or RTA treated for 72 hours to detect the complexes among the chaperone BiP and its client misfolded proteins. The 72-KDa BiP monomer is indicated by the arrow. Unbalance of ER proteostasis upon treatment with RTA determined accumulation of misfolded proteins bound by BiP, indicated as BiP complexes. (E) ML-2 cells, treated for 72 hours as in Figure 1A in the presence or not of 20 mM NAC or of 2.5 mM sodium-4-phenylbutyrate (PBA), were analyzed for propidium iodide (PI) uptake to evaluate cell death (NAC, n = 4 ± SEM; PBA, n = 3 ± SEM). Student t test *P < .05, ****P < .0001. (F) Western blot of protein extracts from ML-2 cells, treated as in panel A, to detect the BiP misfolded protein complexes. NAC relieved oxidative stress induced by RTA and rescued the functionality of the ER, as indicated by the reduction of BiP protein level and by the loss of BiP complexes. A similar effect, although in minor measure, was achieved by PBA.
The clinical outcome of FLT3-ITD+ AML and the strong evidence of the leukemogenic role of mutant FLT3 promoted the development of tyrosine kinase inhibitors (TKIs).13 Clinical trials with TKIs, both as monotherapy and in combination with chemotherapy, resulted in incomplete responses and insurgency of resistance.14,15 Different strategies to target FLT3-ITD have been explored and are related to FLT3-ITD structural defects or specific pathways activated by its aberrant signaling. The proteasome inhibitor bortezomib determined autophagy-mediated FLT3-ITD degradation and cell death of FLT3-ITD+ AML cells16 ; inhibition of FLT3-ITD glycosylation by Tm caused increased ER stress and cell death and acted in synergy with a TKI17 ; pharmacological induction of oxidative stress enhanced the efficacy of the TKI18 ; RA synergized with FLT3-TKI to eliminate leukemia stem cells19 ; eventually, a combination of RA and ATO on FLT3-ITD+ AML cell lines inhibited FLT3-ITD signaling, causing cell death.20 Altogether, these studies indicate the high interest of the scientific community in identifying a combination of drugs able to target the leukemogenic mutation FLT3-ITD.
Here, we demonstrate that the RTA combination efficiently eliminated AML cells with diverse genetic backgrounds, such as the ML-2 and MV-4-11 cell lines, and primary cells from patients FLT3-ITD+ disease with different additional mutations. An important novelty of this work is that in combining RA, Tm, and ATO, we could use low doses of each drug, which had little or no effect when used as single agents, maximizing synergy and possibly reducing toxicity. Although our results need to be further validated in a wider patient cohort and in in vivo models, they provide a proof of concept that low amounts of drugs that generate ER and oxidative stresses combined with RA could be an effective targeted therapy to hit AML cells characterized by MLL fusion proteins and FLT3-ITD mutation.
Acknowledgments
The authors acknowledge Fabrizio Padula for technical assistance.
The research leading to these results was funded by the Associazione Italiana per la Ricerca sul Cancro (AIRC) investigator grant (IG) 2018–ID 21406 project, the Istituto Pasteur Italia–Fondazione Cenci Bolognetti “Call 2018” and “Progetti Ateneo” Sapienza University of Rome (F.F.), the AIRC IG 2015–ID 17352 project (A.F.), the Gruppo Italiano Malattie Ematologiche dell’Adulto (GIMEMA) Foundation “Fund for Ideas 2018” grant (T.O.), and the AIRC 5×1000 call “Metastatic disease: the key unmet need in oncology” to the MYNERVA (MYeloid NEoplasms Research Venture AIRC) project 21267.
This manuscript is dedicated to the memory of F.L.C. with whom we had the great honor to share part of his exciting and prestigious scientific life.
Authorship
Contribution: S.M., E.C., F.L., and M.Ś. performed experiments and contributed to the experimental design; T.O., M.D., S.L., S.T., and N.I.N. characterized and provided leukemic bone marrow samples; A.P. characterized and provided healthy bone marrow samples; V.P., L.T., and A.F. contributed to the experimental design and critical reading of the manuscript; S.M., M.T.V., F.L.C., and F.F. planned the research strategy and wrote the paper; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: F.L.C. declares honoraria from and advisory role for Lundbeck. The remaining authors declare no competing financial interests.
Francesco Lo Coco died on 3 March 2019.
Correspondence: Francesco Fazi, Section of Histology & Medical Embryology, Department of Anatomical, Histological, Forensic & Orthopedic Sciences, Sapienza University of Rome, Via A. Scarpa 14-16, 00161 Rome, Italy; e-mail: francesco.fazi@uniroma1.it; and Silvia Masciarelli, Institute of Histology and Embryology, Catholic University of the Sacred Heart, Largo F. Vito 1, 00168 Rome, Italy; e-mail: silvia.masciarelli@unicatt.it.