Key Points
Stem cell mobilization with G-CSF promotes IL-17A secretion by donor CD8+ MAIT cells.
Tbet and RORγt coexpression identifies potential IL-17A–secreting proinflammatory populations after allogeneic stem cell transplantation.
Introduction
Granulocyte colony-stimulating factor (G-CSF)–mobilized peripheral blood (PB) dominates as the stem cell source in clinical transplantation and has increased the incidence of chronic graft-versus-host disease (GVHD).1 Preclinical studies to date suggest a pathogenic role for donor-derived interleukin-17A (IL-17A) in chronic GVHD in the skin and lung (reviewed in MacDonald et al2 ), consistent with human data demonstrating elevated IL-17A levels systemically late after stem cell transplantation (SCT).3 However, it is clear that several nonconventional cell types can secrete IL-17A, including innate immune cells such as γδ T-cells, type 3 innate lymphoid cells, and mucosa-associated invariant T (MAIT) cells.4,5 In the SCT setting, the contribution of these innate donor T-cell populations to IL-17A production and GVHD has yet to be elucidated.
MAIT cells are a relatively recently defined T-cell population shown to produce proinflammatory cytokines, including interferon γ (IFN-γ), tumor necrosis factor, and IL-17A5-7 in response to microbial-derived riboflavin derivatives loaded onto the nonclassical major histocompatibility complex-I-like molecule MR1.8-10 We and others have shown that MAIT cells can have regulatory functions via the promotion of mucosal integrity and microbiome diversity.11-17 MAIT cells are abundant in humans, representing ∼5% of total PB T cells, 10% of CD8 T cells, and up to 45% of liver T cells.5,7 Several studies report that pathogenic donor CD8+ T cells18 or inflammatory donor Tc17 subsets drive GVHD,19-21 but the distinction between IL-17–secreting MAIT cells and the Tc17 subset has not been comprehensively examined and thus the contribution of MAIT cells to IL-17A production in donor grafts has not been defined. We therefore undertook studies to directly examine human MAIT cells in the PB of healthy donors and allogeneic SCT recipients.
Methods
Human subjects, G-CSF mobilization, and blood collection
Human ethics approval was obtained from the QIMR Berghofer and Royal Brisbane Women’s Hospital human ethics committees with voluntary written informed consent from participating subjects in accordance with the criteria set by the Declaration of Helsinki. Donors were treated with G-CSF (Neupogen) at 10 µg/kg per day for 4 consecutive days with PB collected before and after G-CSF administration. Posttransplant blood samples were collected on days +30 and +180. Donor median age was 52 years (range, 22-65 years); 59% of donors were male and 41% were female. Recipient clinical characteristics are detailed in Table 1.
PBMC isolation, FACS sorting, and cell culture
PB mononuclear cells (PBMCs) were isolated by Ficoll-Paque centrifugation. MAIT (CD3+γδTCRnegCD161+Vα7.2TCR+CD8+CD4neg), CD4Tcon (CD3+γδTCRnegCD161neg/+ Vα7.2TCRnegCD4+CD8neg), and CD8Tcon (CD3+γδTCRnegCD161neg/+Vα7.2TCRnegCD8+ CD4neg) cells were purified from PBMCs by fluorescence-activated cell sorter (FACS) sorting. Equal cell numbers were cultured per well in Iscove modified Dulbecco medium with phorbol 12-myristate 13-acetate (50 ng/mL) and ionomycin (1 µg/mL) for 18 to 24 hours (brefeldin A was added in the final 4 hours). Plasma cytokine levels were measured using human BD CBA Flex sets as described elsewhere.3
Flow cytometry
Fixation and permeabilization were undertaken for intracellular cytokine staining (BD Fix/Perm kit; BD Biosciences) and nuclear staining (Fix/Perm kit; eBioscience) according to the manufacturer’s instructions. Viable cells were identified using the LIVE/DEAD Fixable aqua dead cell staining kit (Invitrogen). Human monoclonal antibodies (mAbs) were purchased from BioLegend (CD3 [UCHT1/HIT3α], CD8α [RPA-T8], CD161 [HP-3G10], Vα7.2TCR [3C10], γδTCR [B1], IFN-γ [4S.B3], IL-17A [BL168], tumor necrosis factor [MAb11], IL-4 [8D4-8], Tbet [4B1O]), BD Biosciences (CD4 [RPA-T4] and CD8α [RPA-T8]), and eBioscience (RORγt [AFKJS-9]). CD1d tetramer was kindly provided by Prof D Godfrey (University of Melbourne). Samples were acquired on a BD LSR Fortessa using BD FACSDiva and analyzed using FlowJo software.
Statistical analysis
Data were analyzed using the paired Wilcoxon signed rank test or the Mann-Whitney U test, where appropriate. P < .05 was considered statistically significant.
Results and discussion
G-CSF mobilization of donors promotes IL-17A secretion from MAIT cells
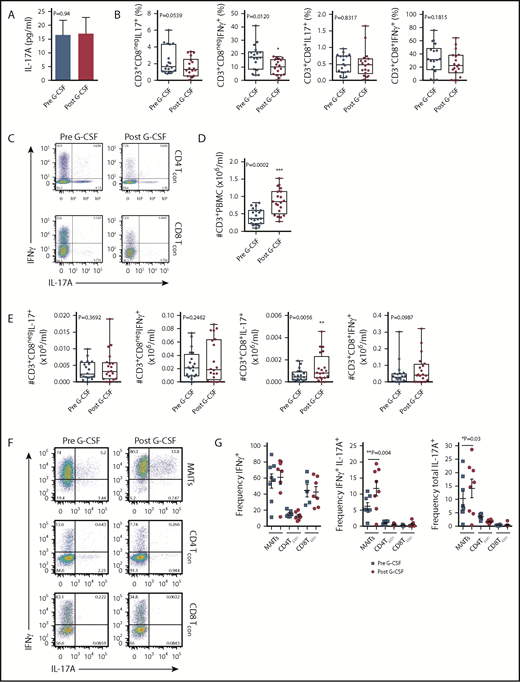
Given our previous findings, which showed elevated levels of plasma IL-17A in SCT recipients late posttransplant,3 we hypothesized that the progeny of lymphoid subsets within the donor PB graft were the likely source of this cytokine. Initially, we examined the IL-17A levels in plasma isolated from the PB of donors administered with G-CSF. While no differences in IL-17A levels were noted with G-CSF administration (Figure 1A), systemic levels were low. We next examined the frequency of IL-17A– and IFN-γ–expressing conventional T cells (Tcon) in stimulated PBMCs isolated from the same donors. In this setting, the proportion of the Th1 subset (here defined as CD3+CD8negIFN-γ+, since CD4 expression was lost on restimulation) was reduced with G-CSF mobilization (Figure 1B-C), while the proportion of the Th17 subset was equivalent (Figure 1B-C). There was no difference in the proportion of Tc1 (CD3+CD8+IFN-γ+) or Tc17 (CD3+CD8+IL-17A+) subsets with G-CSF mobilization (Figure 1B-C). Interestingly, stem cell mobilization with G-CSF resulted in an increase in the total number of CD3+ T cells in the PB (Figure 1D), an effect that directly influences the numbers of T-cell subsets collected in the graft following apheresis.22 Importantly, when the total numbers of T cells were analyzed, only the Tc17 subset was altered and increased significantly (Figure 1E). No differences in the proportion or number of IL-4– and IL-10–producing T cells were observed (data not shown). It is important to note that the proportion of CD8 T cells in whole PB secreting IL-17A was very low (<1%), confirming that the lineage involved was a minor proportion of circulating cells.

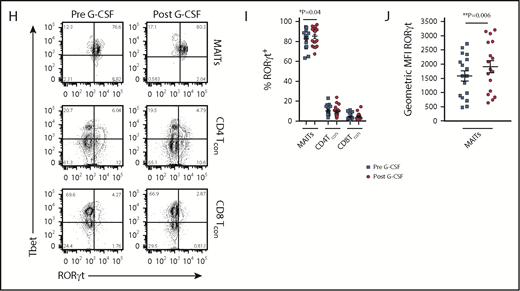
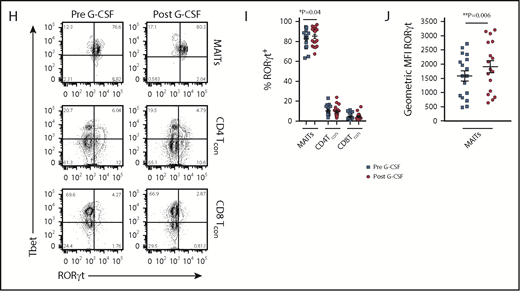
Blood MAIT cells are modified by G-CSF mobilization. (A) Plasma IL-17A levels before and after G-CSF administration (n = 17 donors). (B-C) Frequency and representative FACS plots of Th17 (CD3+CD8negIL-17A+), Th1 (CD3+CD8negIFN-γ+), Tc17 (CD3+CD8+IL-17A+), and Tc1 (CD3+CD8+IFN-γ+) subsets in PBMCs (n = 15 donors). (D) Number of CD3+ T cells per milliliter PB (n = 20 donors); ***P = .0007. (E) Number of Th17 (CD3+CD8negIL-17A+), Th1 (CD3+CD8negIFN-γ+), Tc17 (CD3+CD8+IL-17A+), and Tc1 (CD3+CD8+IFN-γ+) subsets per milliliter PB (n = 15 donors); **P = .0079, Tc17 number before vs after G-CSF. Data were analyzed using the paired Wilcoxon signed rank test and are presented using box-and-whisker plots showing the median with 25th percentiles (whiskers represent minimum to maximum values). (F) Representative FACS plots depicting IFN-γ and IL-17 expression in sorted donor MAIT, CD4Tcon, and CD8Tcon populations stimulated ex vivo with phorbol 12-myristate 13-acetate/ionomycin. (G) Frequency of IFN-γ–, IFN-γ/IL-17A–, and total IL-17A–expressing MAIT, CD4Tcon, and CD8Tcon populations (n = 5-8 donors); **P=.004, frequency of IFN-γ/IL-17A–expressing MAIT cells before vs after G-CSF; *P=.03, frequency of total IL-17A–expressing MAIT cells before vs after G-CSF. (H) Representative FACS plots showing Tbet and RORγt expression by donor MAIT, CD4Tcon, and CD8Tcon populations in unfractionated PBMCs. (I) Frequency of RORγt-expressing MAIT, CD4Tcon, and CD8Tcon populations (n = 17 donors); *P = 0.04. (J) Geometric MFI of RORγt expression in donor MAIT cells; **P = 0.006. Data are presented as mean ± standard error of the mean.
Blood MAIT cells are modified by G-CSF mobilization. (A) Plasma IL-17A levels before and after G-CSF administration (n = 17 donors). (B-C) Frequency and representative FACS plots of Th17 (CD3+CD8negIL-17A+), Th1 (CD3+CD8negIFN-γ+), Tc17 (CD3+CD8+IL-17A+), and Tc1 (CD3+CD8+IFN-γ+) subsets in PBMCs (n = 15 donors). (D) Number of CD3+ T cells per milliliter PB (n = 20 donors); ***P = .0007. (E) Number of Th17 (CD3+CD8negIL-17A+), Th1 (CD3+CD8negIFN-γ+), Tc17 (CD3+CD8+IL-17A+), and Tc1 (CD3+CD8+IFN-γ+) subsets per milliliter PB (n = 15 donors); **P = .0079, Tc17 number before vs after G-CSF. Data were analyzed using the paired Wilcoxon signed rank test and are presented using box-and-whisker plots showing the median with 25th percentiles (whiskers represent minimum to maximum values). (F) Representative FACS plots depicting IFN-γ and IL-17 expression in sorted donor MAIT, CD4Tcon, and CD8Tcon populations stimulated ex vivo with phorbol 12-myristate 13-acetate/ionomycin. (G) Frequency of IFN-γ–, IFN-γ/IL-17A–, and total IL-17A–expressing MAIT, CD4Tcon, and CD8Tcon populations (n = 5-8 donors); **P=.004, frequency of IFN-γ/IL-17A–expressing MAIT cells before vs after G-CSF; *P=.03, frequency of total IL-17A–expressing MAIT cells before vs after G-CSF. (H) Representative FACS plots showing Tbet and RORγt expression by donor MAIT, CD4Tcon, and CD8Tcon populations in unfractionated PBMCs. (I) Frequency of RORγt-expressing MAIT, CD4Tcon, and CD8Tcon populations (n = 17 donors); *P = 0.04. (J) Geometric MFI of RORγt expression in donor MAIT cells; **P = 0.006. Data are presented as mean ± standard error of the mean.
We next undertook experiments to directly examine the proinflammatory cytokine expression by MAIT cells. Given the small starting population, and to exclude any potential stimulatory/inhibitory contributions by myeloid populations present in PBMCs during ex vivo culture, we FACS-sorted MAIT cells, CD4Tcon, and CD8Tcon populations and cultured these individually. Strikingly, this revealed MAIT cells were the only CD8+ IL-17A–secreting T-cell subset following G-CSF mobilization (Figure 1F-G), and IL-17A+ MAIT cells coexpressed IFN-γ. Importantly, the proportion of IFN-γ+/IL-17A+–expressing MAIT cells and total IL-17A+–expressing MAIT cells was significantly increased after G-CSF mobilization (Figure 1G). Consistently, analysis of RORγt and Tbet expression in unfractionated PBMCs revealed the proportion of RORγt-expressing MAIT cells was dramatically greater than that of CD4Tcon or CD8Tcon, and this was enhanced further with G-CSF mobilization (Figure 1H-I). Concomitantly, the mean fluorescence intensity (MFI) of RORγt expression in MAIT cells was also elevated after G-CSF mobilization (Figure 1J).
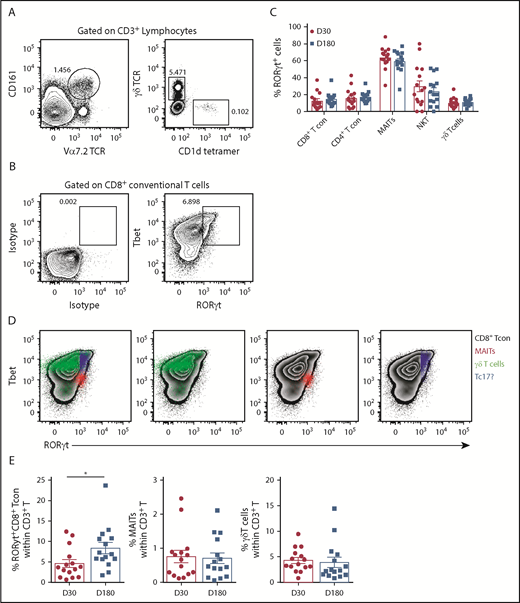
Since IL-17A is not a stable phenotypic marker of Th17/Tc17 cells after SCT, we used RORγt expression, where this is the case.20,23 Analysis of RORγt expression in T-cell subsets (CD4Tcon, CD8Tcon, MAIT cells, natural killer T, and γδT, as detailed in Figure 2A) revealed that conventional CD8 T cells included a Tbet/RORγthi population that was similar to the previously described inflammatory Tc17 cell20 (Figure 2B). Importantly, unlike other T-cell subsets, MAIT cells were predominantly RORγt positive after transplant, with the frequency of RORγt expression remaining unaltered between day +30 and day +180, as was also the case for the other T-cell subsets (Figure 2C). Interestingly, comparison of the MFI of RORγt expression within the total CD8+ T-cell compartment posttransplant showed that MAIT cells (red) displayed an MFI similar to the putative Tbet+RORγt+ Tc17 subset (blue), suggesting a potential capacity for high IL-17A and IFN-γ secretion posttransplant that is known to be a pathogenic signature (Figure 2D). Moreover, Tc17 cells accumulated after SCT in the period when chronic GVHD manifests, while MAIT cells did not (Figure 2E). Unfortunately, we did not have sufficient numbers to adequately power any interrogation of IL-17A–secreting populations relative to chronic GVHD.
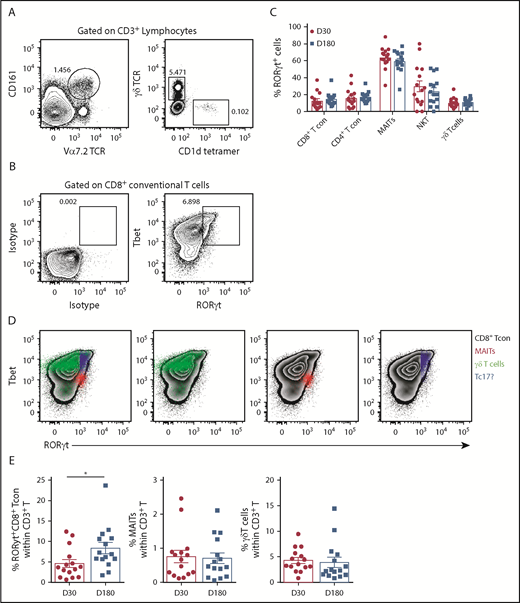
RORγt expression identifies MAIT cells as the predominate potential IL-17A–producing population after allogeneic SCT. (A) Gating strategy employed to identify MAIT (CD3+CD161+Vα7.2TCR+), natural killer T (CD3+CD1d tetramer+), and γδT (CD3+γδTCR+) cells in unfractionated recipient PBMCs. (B) Representative contour plots generated by gating on the total CD8+ T-cell population depicting RORγt and Tbet expression compared with isotype control mAb–stained PBMCs. (C) Frequency of RORγt expression within T-cell populations derived from unfractionated recipient PBMCs at days +30 (n = 15) and +180 (n = 15) posttransplant. (D) Representative contour plots generated by gating on the total CD8+ T-cell population depicting RORγt and Tbet expression in unfractionated PBMCs. RORγt and Tbet expression by MAIT (red), γδT (green), and putative Tc17 (blue) cells is shown. (E) Frequency of RORγthi Tc17, MAIT, and γδT-cells at day +30 (n = 15) and +180 (n = 15) posttransplant. Data presented as mean ± standard error of the mean. *P = .02. TCR, T-cell receptor.
RORγt expression identifies MAIT cells as the predominate potential IL-17A–producing population after allogeneic SCT. (A) Gating strategy employed to identify MAIT (CD3+CD161+Vα7.2TCR+), natural killer T (CD3+CD1d tetramer+), and γδT (CD3+γδTCR+) cells in unfractionated recipient PBMCs. (B) Representative contour plots generated by gating on the total CD8+ T-cell population depicting RORγt and Tbet expression compared with isotype control mAb–stained PBMCs. (C) Frequency of RORγt expression within T-cell populations derived from unfractionated recipient PBMCs at days +30 (n = 15) and +180 (n = 15) posttransplant. (D) Representative contour plots generated by gating on the total CD8+ T-cell population depicting RORγt and Tbet expression in unfractionated PBMCs. RORγt and Tbet expression by MAIT (red), γδT (green), and putative Tc17 (blue) cells is shown. (E) Frequency of RORγthi Tc17, MAIT, and γδT-cells at day +30 (n = 15) and +180 (n = 15) posttransplant. Data presented as mean ± standard error of the mean. *P = .02. TCR, T-cell receptor.
In summary, we show that MAIT cells are the major IL-17A–secreting population within the CD8+ T-cell compartment in humans and that a putative Tc17 population accumulates after SCT. Importantly, we demonstrate that IFN-γ/IL-17A–coexpressing MAIT cells, a phenotype associated with chronic GVHD,19,24 are increased following G-CSF mobilization. Thus MAIT cells are likely mobilized from mucosal tissue by mechanisms similar to stem cells from marrow with enhanced IL-17A secretion in response to expanded myeloid populations secreting permissive cytokines (eg, IL-12/IL-6–secreting monocytes). This would be consistent with recent findings showing human MAIT cells recirculate via lymph by tissue egress.25 Our findings highlight the effects of G-CSF mobilization on MAIT-cell frequency and function. A recent study has demonstrated a correlation between donor MAIT cell numbers in the blood and gastrointestinal tract microbiome constituents known to be associated with protection from acute GVHD.17 We thus suggest that large, adequately powered, clinical studies are needed to correlate known IL-17A–secreting cellular populations within donor grafts and in blood and tissue after SCT, with the subsequent development of GVHD.
Acknowledgments
The authors thank Judy Avery, Justine Leach, Elise Sturgeon, and Nienke Zomerdijk at The Royal Brisbane and Women’s Hospital for coordination/collection of blood samples. They also thank staff in the Flow Cytometry Facility at QIMR Berghofer for cell sorting.
This work was supported by the National Health and Medical Research Council and Queensland Health, Australia (G.R.H.).
Authorship
Contribution: A.V. designed and performed experiments, analyzed data, and wrote the manuscript; K.H.G. designed and performed experiments, analyzed data, and provided helpful discussion; A.N.W., S.D.O., and L.D.S. performed experiments and/or analyzed data; S.-K.T. and G.R.H. enrolled donors and/or patients; K.P.A.M. provided intellectual input and reviewed the manuscript; and G.R.H. provided intellectual input and helped write the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Geoffrey R. Hill, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, Seattle, WA 98109; e-mail: grhill@fredhutch.org; and Antiopi Varelias, QIMR Berghofer Medical Research Institute, 300 Herston Rd, Herston, QLD 4006, Australia; e-mail: antiopi.varelias@qimrberghofer.edu.au.