

Key Points
During a systemic inflammatory response, cell-free DNA is first released by hematopoietic cells and thereafter by nonhematopoietic cells.
Introduction
Systemic inflammation is associated with the release of DNA (cell-free DNA [cfDNA]) into the circulation, the extent of which is proportional to the severity of the systemic inflammation. For example, the plasma concentrations of nucleosomes, complexes consisting of DNA and histones, are significantly increased in sepsis patients.1 Furthermore, in meningococcal sepsis, nonsurvivors have significantly higher nucleosome levels compared with survivors.2 Likewise, the detection of cfDNA in the circulation relates to the severity and outcome of sepsis.3-5
There is an ongoing debate on the cellular source of cfDNA in systemic inflammatory condition; it is often used as a biomarker for neutrophil extracellular trap (NET) formation.6-9 Moreover, it has been argued that, during infection, circulating histones and DNA are mainly released by activated neutrophils in the process of NETosis.10,11 In contrast, others hypothesize that, in sepsis, nucleosomes can also be derived from cell types other than neutrophils.12,13 Widespread cell death of lymphocytes and endothelial cells during sepsis may contribute to extracellular release of DNA and histones.14-16 Organ dysfunction characterizing sepsis resulting from parenchymal and stromal cell death may also lead to the release of cfDNA. Thus, it is currently not clear to what extent neutrophils and other cells contribute to cfDNA in the circulation. The objective of this study was to determine the compartmental source of cfDNA in the circulation during severe systemic inflammation.
Methods
Endotoxemia was induced in 8 healthy, nonsmoking Caucasian male volunteers by an IV injection with Escherichia coli lipopolysaccharide (LPS; US standard reference endotoxin, kindly provided by the National Institutes of Health, Bethesda, MD) at 4 ng/kg body weight. Blood was obtained t = −3, 0, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, and 21 hours relative to LPS injection. Plasma samples of sepsis patients, described elsewhere, were obtained at admission and thereafter daily until day 4.17
In wild-type and bone marrow (BM) chimeric mice, endotoxemia was intraperitoneally induced by injection with E coli LPS (10 mg/kg in 200 μL 0.9% NaCl; L4130 0111:B4, Sigma-Aldrich).
All experiments were approved by the institutional Medical Ethics Committee or the Animal Care and Use Committee (for further details, see supplemental Methods).
Results and discussion
During systemic inflammation, increased amounts of cfDNA can be measured in the circulation using quantitative polymerase chain reaction (qPCR) or in the form of nucleosomes using an enzyme-linked immunosorbent assay.3,18 To obtain a first insight in the origin of cfDNA, we measured nucleosomes in healthy subjects challenged IV with a low dose of LPS. As expected, LPS challenge induced transient systemic inflammation, exemplified by fever, an acute phase response, and an increase in the plasma levels of cytokines and chemokines (data not shown).19 Both nucleosomes and elastase-α1-antitrypsin (EA) complexes, as a measure for neutrophil degranulation, increased, reaching maximum levels at 3 hours after LPS administration (Figure 1A). Furthermore, these levels significantly correlated during LPS challenge (r = 0.79; P < .0001) (Figure 1B). Myeloperoxidase (MPO), another measure for neutrophil activation, increased similarly to the levels of nucleosomes and EA complexes (Figure 1A). However, the contraction phase of MPO differs from that of nucleosomes and EA complexes, which might be explained by a different half-life. These results combined suggest that during a mild, transient inflammatory response, nucleosomes are predominantly released from neutrophils.
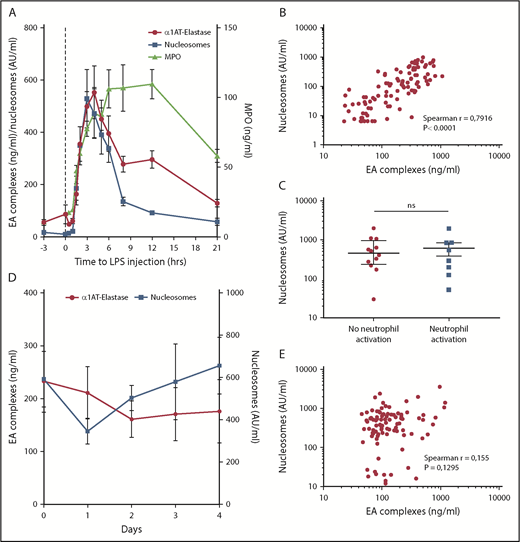
Nucleosomes have other origins besides neutrophils. (A) Nucleosomes and EA complex levels in plasma of humans injected with LPS at t = 0 (n = 8). (B) Nucleosome levels positively correlate to EA complex levels in LPS-challenged subjects. (C) Nucleosome levels in septic patients with neutrophil activation on admission (>100 ng/mL, n = 8) or without neutrophil activation (<100 ng/mL, n = 12) are not statistically different. (D) Nucleosomes and EA complex levels in plasma of sepsis patients (n = 20) at start of admission and during admission to the hospital. (E) No correlation exists between nucleosome and EA complex levels in the previously mentioned sepsis patients. Data are represented as mean ± standard error (A,D) or median ± interquartile range (C). P < .05 was considered significant. ns, not significant.
Nucleosomes have other origins besides neutrophils. (A) Nucleosomes and EA complex levels in plasma of humans injected with LPS at t = 0 (n = 8). (B) Nucleosome levels positively correlate to EA complex levels in LPS-challenged subjects. (C) Nucleosome levels in septic patients with neutrophil activation on admission (>100 ng/mL, n = 8) or without neutrophil activation (<100 ng/mL, n = 12) are not statistically different. (D) Nucleosomes and EA complex levels in plasma of sepsis patients (n = 20) at start of admission and during admission to the hospital. (E) No correlation exists between nucleosome and EA complex levels in the previously mentioned sepsis patients. Data are represented as mean ± standard error (A,D) or median ± interquartile range (C). P < .05 was considered significant. ns, not significant.
In a next step, we measured nucleosomes and neutrophil activation in patients with severe systemic inflammation caused by sepsis (n = 20). Characteristics are given in supplemental Table 1. There was no difference in nucleosome levels between patients with or without enhanced neutrophil degranulation (EA value >100 ng/mL20 ) on admission (Figure 1C). The courses of EA and nucleosomes during follow-up did not show any overlap (Figure 1D) and there was no correlation between EA and nucleosomes levels during hospitalization (r = 0.155; P = .13; Figure 1E). Thus, in patients with sepsis, there was no obvious correlation between neutrophil degranulation and cfDNA, indicating other potential cellular sources of cfDNA than neutrophils. Indeed, in various samples of septic patients, cellular activation and cell death markers of other cell types are increased, making it likely that these cells also provide a source for cfDNA (supplemental Table 2).
To investigate whether nonhematopoietic cells add to the total cfDNA in severe systemic inflammation, we used an endotoxemia mouse model. After LPS injection, both nucleosome and MPO levels increased, peaking at 24 hours (Figure 2A-B). Although nucleosomes and MPO correlated significantly (r = 0.77; P < .0001; supplemental Figure 1A), the ratio between nucleosomes and MPO increased over time (Figure 2C), suggesting that a nonneutrophil source adds to the total amount of nucleosomes. Furthermore, together with increased nucleosome levels (supplemental Figure 1B), we detected an increase of sVCAM-1 (supplemental Figure 1C) and sE-Selectin (supplemental Figure 1D) as measurements of endothelial activation after LPS challenge.
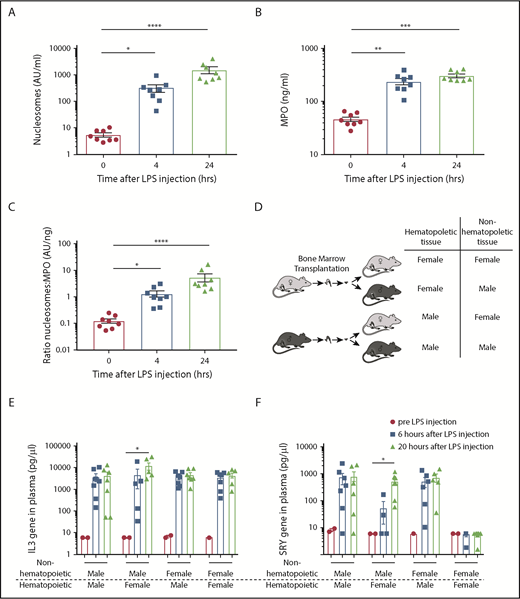
Upon LPS challenge in mice, nucleosomes are derived from both hematopoietic and nonhematopoietic cells. Plasma nucleosomes (A) and MPO (B) levels after intraperitoneal injection of LPS in mice; n = 8 per group. (C) The ratio of nucleosomes vs MPO in LPS-challenged mice. (D) Model of male/female mixed bone marrow chimera mice. General IL-3 gene PCR (E) and specific SRY PCR (F) on extracellular DNA before and 6 and 20 hours after LPS challenge in male/female mixed chimera mice. Pre-LPS (●), n = 1-2 per group; 6 hours after LPS (▪), n = 4-7; and 20 hours after LPS (▲), n = 5-7. Data are presented as mean ± standard error. P < .05 was considered significant. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Upon LPS challenge in mice, nucleosomes are derived from both hematopoietic and nonhematopoietic cells. Plasma nucleosomes (A) and MPO (B) levels after intraperitoneal injection of LPS in mice; n = 8 per group. (C) The ratio of nucleosomes vs MPO in LPS-challenged mice. (D) Model of male/female mixed bone marrow chimera mice. General IL-3 gene PCR (E) and specific SRY PCR (F) on extracellular DNA before and 6 and 20 hours after LPS challenge in male/female mixed chimera mice. Pre-LPS (●), n = 1-2 per group; 6 hours after LPS (▪), n = 4-7; and 20 hours after LPS (▲), n = 5-7. Data are presented as mean ± standard error. P < .05 was considered significant. *P < .05, **P < .01, ***P < .001, ****P < .0001.
To further specify the origin of cfDNA during endotoxemia, we performed a sex-mismatched BM transplantation in mice (Figure 2D). After complete hematopoietic recovery (data not shown), the transplanted mice were challenged intraperitoneally with LPS. We identified the compartmental source of the cfDNA, which is either hematopoietic or nonhematopoietic, by using a specific mouse SRY qPCR as a measure for male DNA, and interleukin-3 (IL3) qPCR as a measure for total DNA. The IL-3 PCR detected a comparable amount of cfDNA in the different groups per time point, indicating similar kinetics of cfDNA release in different BM chimeric groups (Figure 2E). In female mice transplanted with male BM, cfDNA could be measured at 6 and 20 hours after LPS challenge using SRY PCR, identifying the hematopoietic compartment as a source of cfDNA both early and late after LPS challenge (Figure 2F). Interestingly, in male mice that had received BM of female mice, cfDNA using SRY PCR was detected at 20 hours but at <6 hours after LPS challenge. Thus, in later stages of severe systemic inflammation, cfDNA is released by both hematopoietic and nonhematopoietic cells, whereas in earlier stages cfDNA, release can be attributed more to hematopoietic cells.
Our results suggest a biphasic release of cfDNA during severe systemic inflammation: first by hematopoietic cells and thereafter by nonhematopoietic cells. Indeed, activation of neutrophils in the form of NETs has been described to induce epithelial and endothelial cell death, and neutrophil activation in sepsis has been associated with organ dysfunction and fatality.1,21,22 Furthermore, we reported that activation of cytotoxic cells such as cytotoxic lymphocytes and natural killer cells contributes to organ dysfunction by apoptosis induction.23 Last, the release of damage-associated molecular patterns resulting from this cell death at later stages may perpetuate systemic inflammation, which might contribute to both hyperinflammation and immunoparalysis in sepsis.24 The immunoparalysis characterized by extensive lymphocyte apoptosis might even shift the balance more toward nonhematopoietic cfDNA, but further research is warranted to confirm this. In this article, we make a distinction between a hematopoietic or nonhematopoietic source of cfDNA. New sequencing techniques such as plasma DNA tissue mapping, using genome-wide methylation sequencing, may be useful to further pinpoint the specific source of cfDNA in humans.25
In conclusion, we propose a biphasic pattern of cfDNA release in systemic inflammation, first by hematopoietic cells and later additionally by nonhematopoietic cells. Understanding the kinetics of cfDNA may provide a point of interference during systemic inflammation.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the patients and healthy volunteers who participated in this study.
Authorship
Contribution: A.J.v.d.M. and S.Z. designed the experiments; A.J.v.d.M., A.K., A.J.H., and A.A.S. performed the experiments and analyzed the results; A.J.v.d.M., A.K., and S.Z. wrote the manuscript; and A.J.H., A.A.S., C.E.v.d.S., W.A.W., C.V., and T.v.d.P. critically reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for A.J.H. is Department of Plasma Proteins, Sanquin Research, Amsterdam, The Netherlands.
Correspondence: Sacha Zeerleder, Department of Hematology and Central Hematology Laboratory, Inselspital, Bern University Hospital, Freiburgstr 3, CH-3010 Bern, Switzerland; e-mail: sacha.zeerleder@insel.ch.


