Abstract
Philadelphia chromosome-like B-cell acute lymphoblastic leukemia (Ph-like ALL) accounts for 15% to 30% of B-cell acute lymphoblastic leukemia in older children, adolescents, and adults and is associated with high rates of conventional treatment failure and relapse. Current clinical trials are assessing the efficacy of the addition of tyrosine kinase inhibitors (TKIs) to chemotherapy for children and adults with Ph-like ALL harboring ABL class translocations or CRLF2 rearrangements and other JAK pathway alterations. However, real-time diagnosis of patients can be quite challenging given the genetic heterogeneity of this disease and the often cytogenetically cryptic nature of Ph-like ALL-associated alterations. In this review, we discuss the complex biologic and clinical features of Ph-like ALL across the age spectrum, available diagnostic testing modalities, and current clinical treatment strategies for these high-risk patients. We further propose a practical and step-wise approach to Ph-like ALL genetic testing to facilitate the identification and allocation of patients to appropriate clinical trials of TKI-based therapies or commercially available drugs. Although the majority of patients with Ph-like ALL can be successfully identified via current clinical assays by the end of induction chemotherapy, increasing diagnostic efficiency and sensitivity and decreasing time to test resulting will facilitate earlier therapeutic intervention and may improve clinical outcomes for these high-risk patients.
Biology and incidence of Ph-like acute lymphoblastic leukemia
The Philadelphia chromosome-like subtype of B-cell acute lymphoblastic leukemia (Ph-like ALL) was initially identified by a characteristic gene expression signature present in patients with very poor clinical outcomes and a high rate of IKZF1 and other B-cell–associated transcription factor deletions.1-3 Similarities in these kinase-activated gene-expression patterns to those of patients with BCR-ABL1–rearranged (Philadelphia chromosome-positive [Ph+]) disease led to this subtype being referred to as “BCR-ABL1–like” or “Ph-like,” which is now recognized as a provisional leukemia subtype in the World Health Organization 2016 classification.4 Although the Ph-like signature itself has been defined somewhat differently by various groups in Europe and North America,1,2,5-7 these leukemias collectively are now known to be driven by a wide variety of gene fusions, insertions/deletions, and truncations involved in kinase and cytokine signaling potentially targetable with tyrosine kinase inhibitors (TKIs).8-11
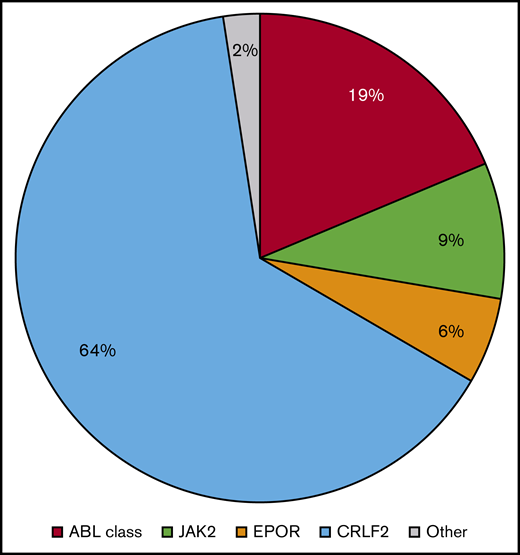
Over the past decade, the most striking finding in the characterization of Ph-like ALL has been its significant genetic heterogeneity with >70 discrete alterations reported to date.11-13 The most common kinase fusions are divided into 4 major categories based upon the presumed 3′ functional fusion partner: ABL class (ABL1 [Abelson kinase 1], ABL2 [Abelson kinase 2], CSF1R [colony-stimulating factor 1 receptor], PDGFRA [platelet-derived growth factor A], and PDGFRB [platelet-derived growth factor B]) rearrangements, CRLF2 (cytokine receptor-like factor 2) rearrangements, EPOR [erythropoietin receptor] rearrangements, and JAK2 rearrangements. The categorization is based upon the similarity of functions of these partners and their potential sensitivity to kinase inhibitors (eg, SRC/ABL/PDGFR inhibitors for ABL class fusions8,14 and JAK inhibitors for CRLF2, EPOR, and JAK2 rearrangements15-18 ). Although the frequency of specific Ph-like kinase fusions varies slightly with age, these major subtypes have been reported across the age spectrum and are universally associated with poor clinical outcomes.8,9,19-25 CRLF2 rearrangements are most common (64.2%), followed by ABL class (18.7%), JAK2 (9.0%), and EPOR (5.7%) rearrangements (Figure 1). Other rare Ph-like ALL–associated alterations (2.4%) have been reported, such as SH2B3 deletions potentially targetable by the JAK inhibitor ruxolitinib and NTRK fusions potentially targetable by the TRK inhibitors crizotinib and larotrectinib. The continued discovery of new lesions emphasizes the critical importance of unbiased genetic testing that is discussed in greater detail in "Ph-like ALL clinical diagnostics." The spectrum of known Ph-like ALL–associated kinase fusions and their relative frequency in children and adults are listed in Table 1, although these numbers likely slightly underestimate all unique rearrangements, given that several fusions involve different exons of the 5′ partner genes. In all of these cases, the resulting oncogenic fusion product is in-frame and retains the functional domains of the 3′ partner gene.

Relative frequency of Ph-like ALL alterations in children, adolescents, and adults. Summary data from 5 recent clinical studies (n = 2506 cases) depict the most common ABL class and CRLF2/JAK pathway–associated translocations occurring in children and adults with Ph-like ALL.8,20,24,44,45
Relative frequency of Ph-like ALL alterations in children, adolescents, and adults. Summary data from 5 recent clinical studies (n = 2506 cases) depict the most common ABL class and CRLF2/JAK pathway–associated translocations occurring in children and adults with Ph-like ALL.8,20,24,44,45
The vast majority of these alterations are cytogenetically cryptic and not readily detected by traditional karyotype analysis, which historically made recognition of Ph-like ALL quite challenging or impossible. The advent of more sophisticated RNA-based molecular fusion assays (and DNA-based next-generation sequencing [NGS] analysis) has greatly facilitated identification of Ph-like ALL–associated alterations with significant discovery of new translocations involving other 5′ fusion partners. Although not all methods of identifying fusions are equivalent, a rough estimate of the total number of unique fusions may be approximated by applying Chao estimators for population richness.26 Based upon the total number of fusions currently identified, the number of those that have been seen exactly once, and the number of those that have been seen exactly twice, an estimate of the probable number of unique fusions may be generated. Application of the Chao 2 estimator to Ph-like ALL yields a predicted value of 192 fusion types if the “Other” category is included and a predicted value of 113 types if only the ABL class, CRLF2, EPOR, and JAK2 rearrangements are considered. As more data are generated and fewer singletons are identified, these estimates will likely improve; however, it seems likely that discovery of Ph-like ALL–associated fusions is only about halfway complete.
As mentioned previously, rearrangement of CRLF2 is the most frequent genetic alteration in Ph-like ALL. CRLF2 alterations are nearly always accompanied by significantly elevated expression of CRLF2 transcripts, either by translocation adjacent to the immunoglobulin heavy chain enhancer [t(X;14) or t(Y;14) resulting in IGH-CRLF2 rearrangement] or by promoter replacement (P2RY8-CRLF2 fusion from intragenic deletion of the pseudoautosomal region of chromosomes X and Y). It should be noted that 5% to 10% of patients with CRLF2-rearranged (CRLF2-R) acute lymphoblastic leukemia (ALL; particularly children with National Cancer Institute [NCI] standard risk disease, defined as <10 years old presenting with white blood cell counts <50 000 cells per microliter27 ) have distinctly different gene-expression profiles without the kinase-activated signature and thus are not considered to have Ph-like disease.
Among NCI high-risk (HR) B-precursor ALL (B-ALL) patients (≥10 years of age and/or with diagnostic white blood cell count ≥50 000 cells per microliter27 ) in the United States, the frequency of IGH-CRLF2 rearrangement appears to be at least twice that of P2RY8-CRLF2 and increases with age.20,22-25 This phenomenon has not necessarily been the case in studies outside of the United States; P2RY8-CRLF2 has been reported to be more common, particularly in children with NCI standard-risk B-ALL who may or may not have the BCR-ABL1–like or Ph-like expression signature.21,28-31 These genetic differences may be attributable in part to the higher prevalence of CRLF2 rearrangements among patients with HR B-ALL of Hispanic/Latino and Native American ancestry, who are more common in North America; however, these discrepancies remain incompletely elucidated. The GATA3 single nucleotide polymorphism risk allele rs3824662, which is associated with an increased risk for leukemia relapse, occurs at a higher frequency in patients with Ph-like ALL, particularly those with CRLF2 rearrangements.32,33
Mutations in JAK genes are highly correlated with CRLF2 rearrangements in Ph-like ALL. Notably, these B-ALL–associated JAK2 mutations (and, occasionally, JAK1 mutations) are distinct from the canonical JAK2 V617F mutation that occurs frequently in adults with myeloproliferative neoplasms.34 Approximately 40% to 50% of cases of CRLF2-R Ph-like ALL harbor pathogenic JAK2 mutations (most commonly R683G in the pseudokinase domain),35,36 which almost always occur only in the setting of CRLF2 rearrangement and have not been seen in conjunction with other Ph-like alterations. Interestingly, ∼50% to 60% of children with Down syndrome–associated ALL have CRLF2 rearrangements (usually P2RY8-CRLF2 fusions) and JAK2 R683G mutations.31,37-42 The frequency of the Ph-like ALL expression signature among patients with CRLF2-R Down syndrome–associated ALL has not been determined.
Some patients with CRLF2-R Ph-like ALL have concomitant IL7R (interleukin-7 receptor) insertions and deletions (indels), which appear to be mutually exclusive from CRLF2-R cases with JAK2 or JAK1 mutations.8,43 Among the 2506 patients with B-ALL in the 5-study summary analysis in Figure 1,8,20,24,44,45 80.8% of identified IL7R indels occurred in patients with the Ph-like subtype (4.3% of all Ph-like patients). It is difficult to estimate the true frequency of IL7R indels in Ph-like ALL because not all studies performed IL7R mutation analysis. Beyond Ph-like ALL, IL7R indels have been reported in 5% to 10% of patients with T-ALL,46 and JAK1 and JAK3 point mutations (but not JAK2) are also relatively common in T-ALL.47-49
Finally, mutations in NRAS, KRAS, PTPN11, NF1, BRAF, and FLT3 have been detected in a small number of patients with Ph-like ALL. Many of these (usually subclonal) Ras pathway mutations arise in conjunction with sentinel Ph-like translocations (eg, ABL class, CRLF2, EPOR, or JAK2 fusions), whereas other mutations appear to occur in isolation.8,9 It is plausible that these mutations could be targeted with MEK or FLT3 inhibitors,50-53 but such strategies have not been formally evaluated in patients with Ph-like ALL.
Ph-like ALL clinical diagnostics
Gene-expression analyses
The first Ph-like classifier was developed over a decade ago by Den Boer and colleagues at Erasmus University and in the Dutch Children’s Oncology Group and used a panel of 110 genes. Hierarchical clustering of the associated probe set expression levels proved highly accurate in separating childhood ALL specimens into 6 distinct categories, including a newly described “BCR-ABL1–like” subtype with a kinase-activated signature very similar to that of Ph+ ALL.9 Two research predictors of the Ph-like ALL expression signature were also developed during this time by researchers in the North America–based Children’s Oncology Group (COG): (1) a predictive analysis of microarrays at the St. Jude Children’s Research Hospital (SJCRH) and (2) a low-density microarray (LDA) analysis at the University of New Mexico.54,55 These 2 modalities have yielded very similar Ph-like ALL classifications of diagnostic SJCRH and COG ALL specimens, although they appear to have somewhat less concordance with methodologies developed in European studies; the discrepancies may be influenced, in part, by the differing ethnic populations in North America vs Europe.21 Although predictive analysis of microarrays and LDA predictors use a continuous scale and apply cutoffs for determining the binary classification of Ph-like or not, the scores themselves are informative about the likelihood of certain fusions.
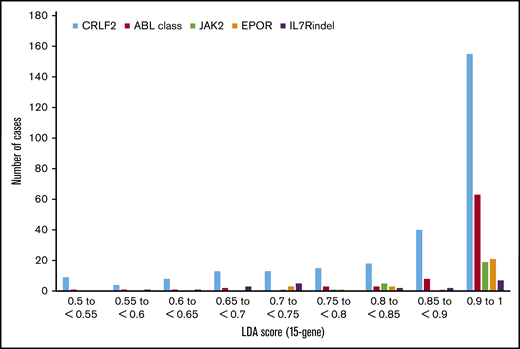
Figure 2 shows results from cumulative LDA analyses of 2506 HR B-ALL patient specimens analyzed in 5 large studies.8,20,24,44,45 Using the now clinically validated 8- or 15-gene (IGJ, SPATS2L, MUC4, CRLF2, CA6, NRXN3, BMPR1B, GPR110, CHN2, SEMA6A, PON2, SLC2A5, S100Z, TP53INP1, IFITM1) LDA predictor threshold ≥ 0.5 (range, 0-1) to define a positive Ph-like signature,5 the majority of ABL class (93.9%), EPOR (100%), and JAK2 (96.3%) fusion events are associated with scores ≥ 0.7. Most Ph-like CRLF2-R ALL cases (87.6%) also have scores ≥ 0.7. The LDA predictor has largely been used by the COG and other cooperative groups as a cost-effective rapid clinical screening tool to determine which HR B-ALL cases require further specific Ph-like molecular characterization for potential clinical trial participation (∼20%) and which do not (∼80%). A high LDA score can provide a rapid index of suspicion within 48 hours of diagnostic sample receipt regarding the likelihood of Ph-like kinase fusions and can alert treating physicians that TKI addition to chemotherapy may be appropriate for a patient if more detailed testing confirms relevant genetic alterations.

Occurrence of Ph-like ALL fusions and correlation with 15-gene LDA predictor score. Distribution of childhood and adult Ph-like ALL cases by 15-gene LDA score and specific Ph-like gene rearrangement.8,24,45 LDA scores ≥0.5 are considered positive for the Ph-like ALL expression signature. The majority of Ph-like cases have LDA scores ≥0.85, particularly those with ABL class, JAK2, and EPOR rearrangements. Some CRLF2-R cases have lower LDA scores in the 0.5 to 0.8 range.
Occurrence of Ph-like ALL fusions and correlation with 15-gene LDA predictor score. Distribution of childhood and adult Ph-like ALL cases by 15-gene LDA score and specific Ph-like gene rearrangement.8,24,45 LDA scores ≥0.5 are considered positive for the Ph-like ALL expression signature. The majority of Ph-like cases have LDA scores ≥0.85, particularly those with ABL class, JAK2, and EPOR rearrangements. Some CRLF2-R cases have lower LDA scores in the 0.5 to 0.8 range.
Cytogenetics and fluorescence in situ hybridization
Conventional cytogenetics analysis and fluorescence in situ hybridization (FISH) studies are routinely performed in the diagnostic evaluation of patients with newly diagnosed ALL; results are usually available in 7 to 10 days. Although karyotypic analysis can identify major structural alterations [eg, t(9;22) resulting in BCR-ABL1 rearrangement in Ph+ ALL], the majority of Ph-like ALL–associated alterations are cytogenetically cryptic. However, clinical break-apart FISH probes have been developed for many of the 3′ genes commonly involved in Ph-like ALL translocations, including ABL1, ABL2, CRLF2, EPOR, JAK2, and PDGFRB (this probe often also detects CSF1R), with rapid result return often within 3 or 4 days. Although FISH analysis often cannot identify the specific 5′ fusion gene partner, abnormal 3′ gene results can provide the first clinical suspicion for ABL class or CRLF2-R/JAK pathway-mutant Ph-like ALL and allocate patients efficiently who require further molecular characterization. Ostensibly, clinical FISH testing with results return within 7 to 10 days of leukemia diagnosis could facilitate earlier therapeutic intervention with JAK inhibitor or ABL/PDGFR inhibitor addition early in induction chemotherapy (as is done for patients with Ph+ ALL56-58 [and NCT03007147]) while completing more detailed molecular analysis to delineate the specific Ph-like rearrangement.
RT-PCR and polymerase chain reaction analyses
Molecular characterization of specific Ph-like ALL kinase fusions can be rapidly accomplished using RNA/complementary DNA–based reverse-transcriptase polymerase chain reaction (RT-PCR) analyses. These targeted assays have a turn-around time of as little as 2 to 3 days and can be “multiplexed” with capabilities for simultaneous testing of multiple kinase fusions. Multiplexed RT-PCR of 39 known Ph-like fusions was an initial approach used by the COG and other consortia for molecular characterization of Ph-like alterations in LDA+ ALL specimens.9,11 However, these RT-PCR assays had significant “false-negative” potential because 5′ and 3′ genes and breakpoints must be known a priori; thus, these assays were often unable to identify kinase fusions with promiscuous breakpoints or previously unknown 5′ partners.18
DNA-based polymerase chain reaction (PCR) assays have been very useful in the detection of common Ph-like ALL–associated mutations, including JAK2 and JAK1 point mutations35,36 and IL7R indels.43 Rarely, CRLF2 F232C point mutations occur in CRLF2-overexpressing ALL cases59,60 ; these mutations seem to be largely independent of the IGH-CRLF2 and P2RY8-CRLF2 rearrangements and can be easily discovered by PCR. Confirmatory clinical Sanger sequencing of all PCR-detected fusions, point mutations, and indels is recommended.
Flow cytometric immunophenotyping
Increased surface thymic stromal lymphopoietin receptor (TSLPR; encoded by CRLF2) staining of ALL blasts, which is readily detectable by flow cytometry, has proven to be highly predictive of IGH-CRLF2 and P2RY8-CRLF2 rearrangements and CRLF2 F232 point mutations in primary Ph-like ALL cells.15 Clinical TSLPR immunophenotyping (now performed as part of routine diagnostic flow cytometry panels23 ) is highly cost effective and can identify patients with probable CRLF2-R B-ALL within 24 hours of specimen acquisition. Several institutions are now routinely incorporating TSLPR flow cytometry into their diagnostic ALL evaluations and/or using it for potential clinical trial screening (M.Y. Konopleva, MD Anderson Cancer Center [MDACC], personal communication; M.J. Borowitz, Johns Hopkins University, personal communication). Confirmatory genetic testing of TSLPR+ specimens by FISH, fusion analysis (described in detail in "NGS platforms"), and/or RT-PCR should be performed to characterize the specific CRLF2 alterations as well as potential JAK and IL7R mutations by PCR analysis, if desired.
NGS platforms
Several commercial and institutional laboratories now offer RNA- and DNA-based hybrid capture or anchored multiplex PCR–based assays for Ph-like ALL characterization, most of which are quite capable of identifying new fusions. Because these assays have become more widely available and cost effective, many institutions and cooperative groups have shifted testing from multiplexed RT-PCR panels to these more comprehensive methodologies. Although some methods for fusion identification are restricted to known gene partners, a more generic approach that also permits discovery of new fusions relies upon priming of the presumed functional 3′ gene partner and extending through the 5′ fusion partners, whether known or novel. The FoundationOne Heme panel is a targeted combined RNA and DNA sequencing method capable of fusion and mutation detection in >400 cancer-related genes.61 The NanoString nCounter digital molecular barcoding platform can identify >200 known leukemia-associated oncogenic fusions.62 The ArcherDX FusionPlex Heme panel is an example of the more generic fusion detection approach; it uses capture-based chemistry that targets 87 hematologic malignancy–associated genes while remaining agnostic to the 5′ fusion partner,63 thereby facilitating potential discovery of novel fusions. All of these platforms require ∼2 to 4 weeks for clinical result reporting, although they have appreciable advantages of more comprehensive and informative analyses.
Whole-exome, transcriptome, and genome sequencing
Rarely, kinase fusions or other Ph-like mutations are not identified in clearly LDA+ ALL specimens. Research-level transcriptomic/RNA sequencing (RNAseq) has been performed on some of these specimens and identified previously unknown alterations that are probable oncogenic drivers in Ph-like ALL.18 Clinical RNAseq is becoming increasingly available and will likely augment or replace current diagnostic targeted testing platforms, although this approach remains quite costly and with relatively slower turnaround time given the complex bioinformatics analysis required. RNAseq analysis can be further complicated by the fact that many fusions are expressed at very low levels, and the fidelity and depth of the sequencing are paramount in being able to distinguish low-level fusions from sequencing artifacts. Whole-exome sequencing was previously used in the initial characterization of Ph-like ALL64 and is useful for point mutation and indel detection, but it generally misses fusions that involve areas outside of coding regions. Whole-genome sequencing (WGS) is another useful technique for characterizing Ph-like ALL, but is currently largely performed only at the research level given its high cost and prolonged timing for test resulting. Although WGS is capable of identifying genomic rearrangements, mutations, and indels, it is incapable of demonstrating that a rearrangement results in a coding fusion or altered transcript that must be identified via RNAseq.
Clinical implementation of Ph-like ALL screening: an example of the current COG approach
LDA screening of all pediatric and adolescent and young adult (AYA) patients with HR B-ALL has been broadly implemented by the COG and used by other consortia to efficiently identify patients with Ph-like ALL who merit additional detailed genetic testing and may be eligible for clinical trials testing relevant TKIs with chemotherapy.11 In practice, LDA results have been returned within 48 to 72 hours, allowing rapid “ruling out” of the 70% to 80% of non-Ph–like ALL patients (“LDA−”) and triggering further genetic testing recommendations for patients with LDA positivity. Of note, the LDA assay also detects specimens with BCR-ABL1 and ETV6-RUNX1 rearrangements due to similarities in expression signatures; accordingly, such patients are not allocated for further testing. In the COG workflow, specimens identified as Ph-like are initially triaged based upon the level of CRLF2 expression (high or low) assessed by LDA, including direct identification of potential P2RY8-CRLF2 fusions in CRLF2-overexpressing specimens. Ph-like specimens with high CRLF2 expression that test negative for the P2RY8-CRLF2 fusion by LDA are then assessed for IGH-CRLF2 translocations by FISH assays, with results returned in 1 week. All CRLF2-R samples are further subjected to JAK1, JAK2, and IL7R PCR mutation analysis that usually also requires 1 week for resulting. LDA+ specimens with normal CRLF2 expression are sent for customized Archer-based kinase fusion testing to assess for JAK2, EPOR, and ABL class rearrangements and other rare Ph-like–associated alterations, with a current turnaround time ∼ 3 weeks.11 As above, clinical RNAseq analysis can be performed for specimens with the Ph-like expression signature in which no kinase fusion or other oncogenic mutation is identified, but this testing often requires 4 to 8 weeks to result and is generally too slow to allow allocation of relevant patients to TKI-based clinical trials that begin at the consolidation phase of therapy.
Current clinical trials for patients with Ph-like ALL
Addition of ABL-targeting TKIs to chemotherapy for patients with Ph+ ALL has markedly improved relapse-free and overall survival and provides an important precision medicine paradigm for other HR leukemia subtypes.56-58,65-67 Analogous approaches for patients with Ph-like ALL are under investigation given similar activated kinase expression profiles and kinase gene fusions that can also be targeted with small molecule inhibitors. The majority of Ph-like ALL cases can be “binned” into 2 classes for therapeutic targeting purposes: ABL class mutant (including ABL1, ABL2, CSF1R, and PDGFRB rearrangements) and JAK pathway mutant (including CRLF2, JAK2, or EPOR rearrangements, SH2B3 deletions, and IL7R indels). As described in detail in "ABL class alterations" and "CRLF2 rearrangements and other JAK pathway alterations," several clinical trials are investigating whether addition of the SRC/ABL/PDGFRB inhibitor dasatinib or the JAK1/2 inhibitor ruxolitinib to multiagent backbone chemotherapy can improve the known poor clinical outcomes of children and adults with Ph-like ALL (Table 2).
ABL class alterations
Several groups have reported potent activity of imatinib or dasatinib in vitro and in vivo in ABL class-mutant Ph-like ALL cells and patient-derived xenograft models.8,14,25 Anecdotal case reports have further demonstrated clinical efficacy of combining these TKIs with chemotherapy in children and AYAs with chemotherapy-refractory B-ALL with PDGFRB, ABL1, or ABL2 fusions.8,68-70 Imatinib, dasatinib, and later-generation TKIs are approved by the US Food and Drug Administration and European Medicines Agency for adults and children with chronic myeloid leukemia or Ph+ ALL, but have not been approved for patients with Ph-like ALL. Multiple clinical trials are now studying nonrandomized addition of dasatinib to chemotherapy in patients with relapsed or newly diagnosed ABL class-mutant Ph-like ALL (Table 2). A phase 1/2 study conducted at MDACC is testing the safety and potential efficacy of dasatinib with hyper-CVAD (cyclophosphamide, vincristine, doxorubicin, dexamethasone) chemotherapy in adolescents and adults with relapsed/refractory Ph-like ALL and ABL class fusions (NCT02420717); interim data analysis demonstrated safety of combination therapy without identified dose-limiting toxicity.71 The potential efficacy of dasatinib and postinduction chemotherapy in children and AYAs with de novo ABL class-mutant Ph-like ALL has been studied descriptively on arms of the phase 3 COG AALL1131 (NCT01406756) and the SJCRH Total XVII (NCT03117751) trials, with comparison with historic control data from Ph-like patients treated with chemotherapy alone. These up-front TKI addition approaches aim to decrease relapse risk and to improve overall survival in children and AYAs with Ph-like ALL, as has been observed in patients with Ph+ ALL treated with imatinib or dasatinib. Although anecdotal case reports have described responses of patients with ABL class Ph-like ALL to imatinib or dasatinib,8,68,69 results from these larger clinical trials are not yet known.
CRLF2 rearrangements and other JAK pathway alterations
Preclinical studies have also shown appreciable activity of JAK inhibitors in vitro and in vivo in CRLF2-R and other JAK pathway-mutant Ph-like ALL cell lines and patient-derived xenograft models.16-18,72-74 Although several JAK inhibitors have been studied in patients with autoimmune diseases and hematologic malignancies,75,76 ruxolitinib (a selective JAK1/JAK2 inhibitor77 ) has been most widely studied in ALL. Ruxolitinib is approved by the US Food and Drug Administration and European Medicines Agency for treatment of adults with myelofibrosis and polycythemia vera (usually harboring somatic JAK2 V617F mutations), but it is not approved for use in children or in patients with acute leukemias. The COG ADVL1011 phase 1 trial established the safety and recommended phase 2 dose of ruxolitinib in pediatric patients (1-21 years of age) with relapsed/refractory solid tumors and hematologic malignances. A maximally tolerated dose of ruxolitinib and major dose-limiting toxicities were not identified in this study.78
The aforementioned MDACC Ph-like ALL phase 1/2 trial (NCT02420717) also tested low-dose ruxolitinib with hyper-CVAD chemotherapy for adolescents and adults with relapsed/refractory CRLF2-R/JAK-mutant ALL. Combination therapy was well tolerated, but limited efficacy was observed.71 Similar to the AALL1131 dasatinib arm for patients with ABL class alterations, the COG is also testing the safety and efficacy of ruxolitinib addition to chemotherapy in children and AYAs with newly diagnosed Ph-like ALL and CRLF2 rearrangements or other JAK pathway alterations via the single-arm AALL1521 phase 2 trial (NCT02723994). Patients are stratified into 4 cohorts by Ph-like genetic alterations (CRLF2-R, CRLF2-R with JAK mutation, other JAK pathway lesions) and by end-induction minimal residual disease status (≥0.01 or <0.01% by flow cytometry) to allow for the more precise study of potential efficacy differences among these subgroups. Safety data for ruxolitinib with postinduction chemotherapy from part 1 of this trial were recently reported, and no dose-limiting toxicity of combination therapy was identified.79 Part 2 of AALL1521 is currently assessing the efficacy of ruxolitinib with chemotherapy and will compare outcomes with those of patients treated with chemotherapy alone on the prior COG AALL0232 phase 3 trial (NCT00075725).80 The SJCRH Total XVII trial is also assessing the safety and potential efficacy of ruxolitinib with chemotherapy in children with de novo CRLF2-R/JAK pathway–mutant Ph-like ALL (NCT03117751). Finally, a recently opened phase 1 trial at the University of Chicago and other institutions (NCT03571321) is studying ruxolitinib in combination with the pediatric-inspired CALBG 10403 chemotherapy regimen (NCT00558519)81,82 specifically in AYA patients (18-39 years of age) with newly diagnosed Ph-like ALL with a planned phase 2 expansion if safety is demonstrated.
Conclusions and diagnostic recommendations
Ph-like ALL is a common leukemia subtype in children and adults that is associated with high rates of chemotherapy resistance and relapse. Historically, clinical diagnosis of patients with Ph-like ALL has proven quite challenging given the now-known significant genetic heterogeneity of associated kinase fusions that are often cytogenetically cryptic and that previously required lengthy step-wise and costly testing that, nonetheless, failed to identify many lesions. Sophisticated RNA-based testing platforms (many of which are far more capable of new fusion partner discovery) that are now widely clinically available have appreciably facilitated identification of patients with Ph-like ALL and their specific leukemia-associated fusions, but these approaches require several weeks for data resulting. Instead, routine clinical FISH testing with the use of new ABL1, ABL2, CRLF2, JAK2, and PDGFRB probes and flow cytometric immunophenotyping for increased TSLPR surface expression may provide early suspicion for Ph-like ALL in relevant patients. Such approaches could be used for early intervention with appropriate TKI addition to chemotherapy while awaiting specific Ph-like ALL molecular analysis by more detailed testing.
Based upon these scientific and clinical advances, we propose a practical, cost-effective, and time-efficient clinical algorithm to identify patients with Ph-like ALL (Figure 3). All pediatric patients with NCI HR B-ALL and adult patients with B-ALL should undergo routine diagnostic flow cytometric immunophenotyping with a panel that includes an anti-TSLPR (CRLF2) antibody.15,83 All patients should also undergo routine cytogenetics and FISH analysis using ABL1, ABL2, CRLF2, EPOR, JAK2, and PDGFRB probes that can rapidly identify probable Ph-like ALL in specimens with detected aberrant signals. Leukemia specimens with identified increased flow cytometric TSLPR staining should be specifically allocated for CRLF2 FISH, particularly in the AYA population, in which IGH-CRLF2 translocations are more common.84 TSLPR+ specimens with negative CRLF2 rearrangement FISH data should be sent for P2RY8-CRLF2 fusion analysis via RT-PCR or fusion panel testing. For complete genetic characterization, CRLF2-R specimens can also be tested via targeted PCR or more comprehensive NGS to assess potential JAK1 and JAK2 mutations, IL7R indels, and associated transcription factor deletions (eg, IKZF1, CDKN2A, CDKN2B, PAX53 ) that are common in Ph-like ALL and may further contribute to prognosis.
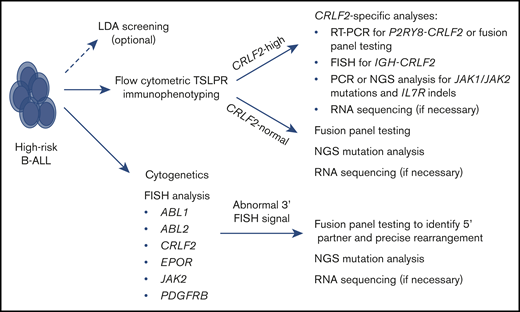
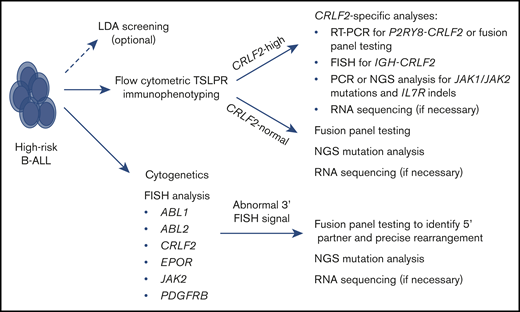
Recommended clinical testing algorithm for identification of patients with Ph-like ALL.
Recommended clinical testing algorithm for identification of patients with Ph-like ALL.
We maintain that adult and HR pediatric B-ALL specimens with normal (nonincreased) TSLPR staining that lack canonical leukemia-associated alterations (eg, BCR-ABL1, ETV6-RUNX1, hyperdiploidy, hypodiploidy, iAMP21, KMT2A rearrangements, TCF3-PBX1, TCF3-HLF) should be sent for comprehensive fusion panel testing and NGS analysis. These testing platforms have rapidly become more affordable and with shorter “turnaround time” for resulting, and the potential discovery of Ph-like (and non-Ph–like) translocations and mutations could have major prognostic and therapeutic implications for patients. It is also plausible that clinical RNAseq analysis will become more widely available and affordable in the near future with more rapid return of results, and such testing may eventually replace targeted cancer fusion and NGS panels. Finally, we propose that LDA analysis remains a helpful screening tool to “rule out” the large number of non-Ph–like cases and to triage selected Ph-like cases for downstream molecular analysis, particularly in the younger pediatric population and those with P2RY8-CRLF2 fusions. Conversely, we suggest that LDA is probably unnecessary for AYA and older patients and those with ABL class, JAK2, EPOR, and IGH-CRLF2 rearrangements that can be readily detected by FISH and fusion analyses and are now well known to be near-universally associated with the Ph-like expression signature and poor clinical outcomes with conventional therapy.
In summary, several clinical trials of dasatinib and ruxolitinib with multiagent chemotherapy are in progress for patients with ABL class-mutant and CRLF2-R/JAK pathway–mutant Ph-like ALL, respectively, but the impact of these interventions upon relapse-free and overall survival is not yet known. Given the remarkable success and clinical tolerability of CD19- and CD22-targeted antibody-based and cellular immunotherapies in patients with relapsed/refractory B-ALL, evaluation of the potential efficacy of combining TKIs with immunotherapy could also be considered in future Ph-like ALL clinical trials. Further refinement of Ph-like ALL testing methodologies with more rapid test resulting will continue to facilitate the swift identification of these extremely HR patients who may benefit from alternative therapeutic approaches and clinical trials.
Acknowledgments
R.C.H. was supported by National Institutes of Health, National Cancer Institute grants R50CA211542 and P30CA118100. S.K.T. was supported by National Institutes of Health, National Cancer Institute grants K08CA18441 and 1U01CA232486; Department of Defense grant CA180683P1; and the Rally Foundation for Childhood Cancer Research.
Authorship
Contribution: R.C.H. and S.K.T. wrote the manuscript.
Conflict-of-interest disclosure: R.C.H. is an inventor on US Patent 8568974 relating to the use of gene-expression signatures to identify Ph-like patients, but receives no royalties. S.K.T. receives research funding from Incyte Corporation.
Correspondence: Sarah K. Tasian, Children’s Hospital of Philadelphia, University of Pennsylvania Perelman School of Medicine, 3501 Civic Center Blvd, CTRB 3010, Philadelphia, PA 19104; e-mail: tasians@email.chop.edu.