Key points
GRK6 regulates the hemostatic response by limiting platelet activation via thrombin and adenosine 5′-diphosphate.
GRK6 regulates the hemostatic response by reducing PAR1/4- and P2Y12-dependent signaling.

Abstract
G protein–coupled receptors (GPCRs) mediate the majority of platelet activation in response to agonists. However, questions remain regarding the mechanisms that provide negative feedback toward activated GPCRs to limit platelet activation and thrombus formation. Here we provide the first evidence that GPCR kinase 6 (GRK6) serves this role in platelets, using GRK6−/− mice generated by CRISPR-Cas9 genome editing to examine the consequences of GRK6 knockout on GPCR-dependent signaling. Hemostatic thrombi formed in GRK6−/− mice are larger than in wild-type (WT) controls during the early stages of thrombus formation, with a rapid increase in platelet accumulation at the site of injury. GRK6−/− platelets have increased platelet activation, but in an agonist-selective manner. Responses to PAR4 agonist or adenosine 5′-diphosphate stimulation in GRK6−/− platelets are increased compared with WT littermates, whereas the response to thromboxane A2 (TxA2) is normal. Underlying these changes in GRK6−/− platelets is an increase in Ca2+ mobilization, Akt activation, and granule secretion. Furthermore, deletion of GRK6 in human MEG-01 cells causes an increase in Ca2+ response and PAR1 surface expression in response to thrombin. Finally, we show that human platelet activation in response to thrombin causes an increase in binding of GRK6 to PAR1, as well as an increase in the phosphorylation of PAR1. Deletion of GRK6 in MEG-01 cells causes a decrease in PAR1 phosphorylation. Taken together, these data show that GRK6 regulates the hemostatic response to injury through PAR- and P2Y12-mediated effects, helping to limit the rate of platelet activation during thrombus growth and prevent inappropriate platelet activation.
Introduction
Platelets play a critical role in hemostasis, thrombosis, and inflammation. Once platelets are recruited to the damaged vessel wall, the primary drivers of platelet activation are thrombin, adenosine 5′-diphosphate (ADP), thromboxane A2 (TxA2), and collagen. With the exception of collagen, each of these agonists signals through G protein–coupled receptors (GPCRs). The G proteins that function as mediators for these receptors are α, β, and γ heterotrimers. GPCRs turn on G proteins by promoting the exchange of GDP for GTP. Gα and Gβγ subunits then dissociate and stimulate their respective effectors. Signaling terminates when Gα hydrolyzes bound GTP back to GDP. Platelet activation can be regulated at multiple places in its signaling network, including at the levels of receptor activation, intracellular Ca2+ elevation, RAP1 activation, and integrin outside-in signaling.1 These different levels of regulatory events are essential to achieve optimal platelet signaling so that platelet activation is neither inadequate (allowing rebleeding to occur) nor overly exuberant (risking vascular occlusion). The first signaling node to control platelet activation after exposure to agonists is at the level of receptor stimulation. In nucleated cells, there are a number of well-described mechanisms for limiting signaling through GPCRs, including receptor phosphorylation, internalization, and shedding. Among them, one of the key regulators of GPCRs is the agonist-dependent phosphorylation by GPCR kinases (GRKs) followed by internalization of receptors.2-4 This process is also known as desensitization, and it attenuates the receptor response in the presence of continuous stimulation of agonists.3 Here we have asked whether GRKs are regulators of platelet activation and, if so, whether they impact the hemostatic response to injury.
The GRK family consists of 7 members (GRK1 to GRK7) that are divided into 3 subfamilies: (1) the rhodopsin kinases, or visual GRKs (numbers 1 and 7); (2) the GRK2/3 family, originally termed β-adrenergic receptor kinases 1 and 2; and (3) the GRK4 family, comprising GRK4, -5, and -6. GRK2, -3, -5, and -6 are ubiquitously expressed. Others show a more tissue-specific distribution.5 GRKs share a relatively conserved kinase domain, an N-terminal Regulator of G protein signaling (RGS) homology domain that is bracketed by a short N-terminal α-helical domain, and a variable C-terminal lipid-binding region.4,6 One of the major differences between RGS proteins and GRKs is that the RGS homology domains of GRKs have little or no GTPase activating protein activity, which is the hallmark of classical RGS proteins. Instead, the ability of GRKs to attenuate signaling is primarily due to receptor phosphorylation.4,7 GRKs play an important role in cardiovascular physiology and the development of cardiac disease.8-11 Changes in GRK expression have been linked to many cardiovascular pathologies, including myocardial infarction, hypertension, and cardiac hypertrophy. A previous report shows that GRK6-deficient mice develop an autoimmune-like disease.12 We have been able to detect GRK2, GRK5, and GRK6 protein expression in both human and mouse platelets, which is consistent with platelet transcriptome13 and proteomics data.14,15 Based on estimated copy number, GRK6 is the form most predominantly expressed in both human and mouse platelets. In a transfected cell–based assay, GRK6 contributes to the desensitization of P2Y12.16 Nothing is known about the impact of GRK6 on platelet activation and its role in modulating the hemostatic response to injury.
In the present study, we show that GRK6 functions as a negative regulator of platelet activation in vitro and the hemostatic response to injury in vivo. Mice lacking GRK6 show an increased rate of platelet deposition during the initial stage of hemostatic thrombus formation after injury. When studied ex vivo, their platelets show an agonist-selective increase in responses to thrombin and ADP, but not to TxA2 or collagen. Finally, the data show that there is increased binding of GRK6 to PAR1 during platelet activation by thrombin. Increased binding of GRK6 is associated with increased phosphorylation of PAR1 during human platelet activation. Based on these observations, we propose that GRK6 helps to establish a stable and controlled hemostatic plug during the early stages of thrombus formation, halting the loss of blood without occlusion of the vessel.
Methods
Mice
The generation of GRK6 knockout mice using the CRISPR-Cas9 genome-editing system was performed as described by Henao-Mejia et al.17 Briefly, zygotes from C57BL/6 mice were injected with Cas9 messenger RNA (100 ng/μL) and guide RNAs (gRNAs) (50 ng/μL). Embryos were then transferred to pseudo-pregnant C57BL/6 female mice. The founder mice were each bred to the F1 generation using C57BL/6 mice for further analysis. All mouse protocols and procedures were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania or Thomas Jefferson University.
Vascular injury
Hemostatic thrombus formation was observed in the cremaster muscle microcirculation of male mice aged 8 to 12 weeks as previously described.18 Briefly, Alexa Fluor 568–labeled anti-CD41 antibody F(ab)2 fragments, Alexa Fluor 647–labeled anti–P-selectin, and Alexa-647 anti-fibrin antibodies were administered via a catheter in the jugular vein. Arterioles 30 to 50 μm in diameter were studied. Vascular injury was induced using a pulsed nitrogen dye laser fired through the microscope objective. Thrombus formation was observed for 3 minutes and analyzed.
Other methods
Material and antibodies, cell culture, platelet preparation, flow cytometry, platelet aggregation, and coprecipitation experiments are described in supplemental Methods.
Statistical analysis
Results are presented as mean ± standard error of the mean (SEM) as shown in the figure legends. Data were analyzed using the Student t test. P ≤ .05 was considered statistically significant.
Results
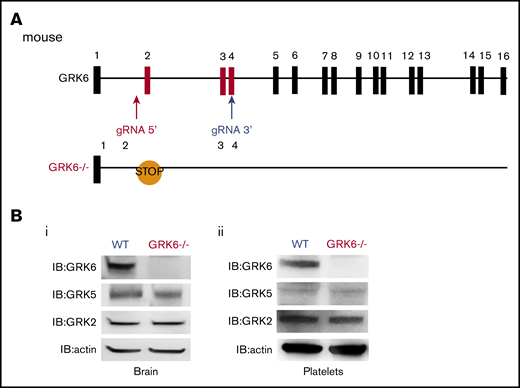
We used CRISPR-Cas9 genome editing to generate GRK6−/− mice by designing gRNAs targeting intron 1 and exon 4 of mouse GRK6 (Figure 1A). This introduced a premature stop codon and also generated a 450-bp deletion between intron 1 and exon 4 (supplemental Figure 1A). The GRK6−/− mice generated by crossing between GRK6+/− mice were born in the expected Mendelian inheritance ratios (supplemental Table 1). The GRK6−/− mice were slightly smaller than their GRK6+/+ littermates, but caught up by 8 to 9 weeks (supplemental Figure 1B). There was ∼10% decrease in platelet counts in GRK6−/− mice. Mean platelet volume, red cell count, and leukocyte count were normal (supplemental Table 2). Deletion of GRK6 did not affect GRK2 or GKR5 protein expression in mouse brain (Figure 1Bi) or in platelets (Figure 1Bii).
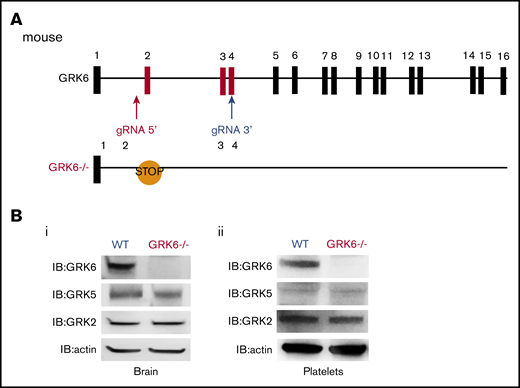
Generation and characterization of GRK6−/−mice. (A) Strategy for introducing stop codon in the mouse GRK6 gene. The gRNA 5′ and gRNA 3′ were combined with Cas9 messenger RNA for subsequent cytoplasmic injection of fertilized mouse eggs. Embryos were then transferred to pseudopregnant C57BL/6 female mice. After birth, 10-day-old mice were tail-snipped, and genomic DNA was extracted for genotyping and sequencing. The founder mice were each bred to the F1 generation using C57BL/6 mice for further analysis. CRISPR-mediated genome editing introduced a premature stop codon in exon 2. (B) Deletion of GRK6 does not affect GRK2 or GRK5 protein expression in mouse brain (i; N = 3) or platelets (ii; N = 2). Lysates were probed for GRK6 before being successively reprobed for GRK5, GRK2, or actin.
Generation and characterization of GRK6−/−mice. (A) Strategy for introducing stop codon in the mouse GRK6 gene. The gRNA 5′ and gRNA 3′ were combined with Cas9 messenger RNA for subsequent cytoplasmic injection of fertilized mouse eggs. Embryos were then transferred to pseudopregnant C57BL/6 female mice. After birth, 10-day-old mice were tail-snipped, and genomic DNA was extracted for genotyping and sequencing. The founder mice were each bred to the F1 generation using C57BL/6 mice for further analysis. CRISPR-mediated genome editing introduced a premature stop codon in exon 2. (B) Deletion of GRK6 does not affect GRK2 or GRK5 protein expression in mouse brain (i; N = 3) or platelets (ii; N = 2). Lysates were probed for GRK6 before being successively reprobed for GRK5, GRK2, or actin.
GRK6−/− mice display an increased initial rate of platelet accumulation in response to injury
Platelet function was studied in vivo with a well-established model that uses a laser to make penetrating injuries in cremaster muscle arterioles as previously described.18,19 The thrombus architecture that forms consists of a core of tightly packed, P-selectin–positive platelets overlaid with a shell of loosely adherent P-selectin–negative platelets. Anti-CD41 is used as a marker for platelet deposition. Anti–P-selectin, which is an α-granule exocytosis marker, is used to distinguish the core from the shell. Fibrin is detected with a fibrin-specific monoclonal antibody that does not recognize fibrinogen.18 Thrombin activity and fibrin deposition in this model are limited to the core.20
Representative images of hemostatic plugs formed in wild-type (WT) and GRK6−/− mice 1 minute after injury are shown in Figure 2Ai. Platelets accumulate more rapidly at the site of injury in GRK6−/− mice than in WT controls (Figure 2Aii). Notably, the slope of platelet accumulation during the early stage of thrombus formation was increased by 2.5-fold in GRK6−/− mice relative to WT controls (Figure 2B). Overall, total platelet accumulation was significantly increased in GRK6−/− mice compared with WT controls (Figure 2Aiii). There was no significant difference in total P-selectin accumulation (Figure 2Aiv). GRK6−/− mice also had normal fibrin accumulation (Figure 2C), indicating that thrombin generation at the site of injury is unaltered in GRK6−/− mice.
![Increased platelet accumulation in response to hemostatic injury. (A) Representative images of hemostatic plugs formed in WT and GRK6−/− mice 1 minute after injury (Ai). (Aii) Confocal intravital fluorescence microscopy was performed to follow platelet accumulation and P-selectin expression. Data are mean ± SEM; 46 injuries in 5 WT mice and 42 injuries in 5 GRK6−/− mice. (Aiii) Graph shows the area under CD41 vs time curve (area under the curve [AUC]). (Aiv) Graph shows the area under P-selectin (+) vs time curve (AUC). The line and error bars show the median and interquartile range. (B) CD41+ area at 45 seconds after injury (Bi). (Bii) The rate of CD41 deposition at 45 seconds after injury, and the bar graph represents the average slope of the liner trend line equation of each injury. Forty-one injuries in 5 WT mice and 41 injuries in 5 GRK6−/− mice. (C) Fibrin deposition after making small penetrating injuries in the cremaster muscle arterioles with laser in GRK6−/− mice and littermate controls. Data are mean ± SEM; 24 injuries in 5 WT mice and 25 injuries in 5 GRK6−/− mice (Ci). (Cii) Bar graphs represent the fibrin accumulation at the end of the 3-minute observation period.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/1/10.1182_bloodadvances.2019000467/3/m_advancesadv2019000467f2.png?Expires=1768076761&Signature=gKQK0h2wFnoJpaWZ4YYfjeu8yoDZD4Z20TthjdtCQxBlKLaDLgPAkoSAubiZx3nveSq-KM3~1qIJgloQNNlULR7WGnwsGU9Eo3k6dlzMnHIt8hfaS6y1zzBQzfu3012eqfa5PPzzx8M7vzXA2g6rEBXXbqifnrzsRmz7Xw8zZlL~Jl6wzBrQJoU9ypHhSqjznEN4OnAG0j5dnRPxY8ZwSRAQzJGWijZBAkwOc3aZxh2SCt2N8xKX0v9cvq~XFXGsoex95FYPI5-JH~-RZNrQSHwajHOjDgoD9YxjufRq5siWRhCcABjGz3Rn7MEAiZf1d8xZ9WRYIcksJKz7WRXgWA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Increased platelet accumulation in response to hemostatic injury. (A) Representative images of hemostatic plugs formed in WT and GRK6−/− mice 1 minute after injury (Ai). (Aii) Confocal intravital fluorescence microscopy was performed to follow platelet accumulation and P-selectin expression. Data are mean ± SEM; 46 injuries in 5 WT mice and 42 injuries in 5 GRK6−/− mice. (Aiii) Graph shows the area under CD41 vs time curve (area under the curve [AUC]). (Aiv) Graph shows the area under P-selectin (+) vs time curve (AUC). The line and error bars show the median and interquartile range. (B) CD41+ area at 45 seconds after injury (Bi). (Bii) The rate of CD41 deposition at 45 seconds after injury, and the bar graph represents the average slope of the liner trend line equation of each injury. Forty-one injuries in 5 WT mice and 41 injuries in 5 GRK6−/− mice. (C) Fibrin deposition after making small penetrating injuries in the cremaster muscle arterioles with laser in GRK6−/− mice and littermate controls. Data are mean ± SEM; 24 injuries in 5 WT mice and 25 injuries in 5 GRK6−/− mice (Ci). (Cii) Bar graphs represent the fibrin accumulation at the end of the 3-minute observation period.
Increased platelet accumulation in response to hemostatic injury. (A) Representative images of hemostatic plugs formed in WT and GRK6−/− mice 1 minute after injury (Ai). (Aii) Confocal intravital fluorescence microscopy was performed to follow platelet accumulation and P-selectin expression. Data are mean ± SEM; 46 injuries in 5 WT mice and 42 injuries in 5 GRK6−/− mice. (Aiii) Graph shows the area under CD41 vs time curve (area under the curve [AUC]). (Aiv) Graph shows the area under P-selectin (+) vs time curve (AUC). The line and error bars show the median and interquartile range. (B) CD41+ area at 45 seconds after injury (Bi). (Bii) The rate of CD41 deposition at 45 seconds after injury, and the bar graph represents the average slope of the liner trend line equation of each injury. Forty-one injuries in 5 WT mice and 41 injuries in 5 GRK6−/− mice. (C) Fibrin deposition after making small penetrating injuries in the cremaster muscle arterioles with laser in GRK6−/− mice and littermate controls. Data are mean ± SEM; 24 injuries in 5 WT mice and 25 injuries in 5 GRK6−/− mice (Ci). (Cii) Bar graphs represent the fibrin accumulation at the end of the 3-minute observation period.
GRK6 deficient platelets have increased platelet activation ex vivo
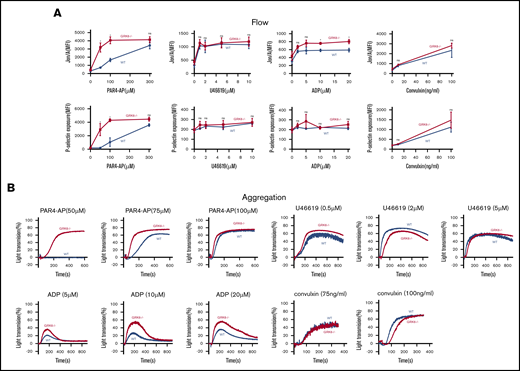
To assess the functional consequences of GRK6 deletion on platelet function, integrin activation and α-granule secretion were compared in platelets from GRK6−/− and GRK6+/+ (WT) littermates using flow cytometry with antibodies specific to activated αIIbβ3 and P-selectin.21 The assays were performed using highly diluted platelet suspensions to minimize signaling induced by secreted mediators such as ADP and TxA2. The results in Figure 3A show that platelet activation with the PAR4 thrombin receptor agonist peptide (PAR4-AP), AYPGKF, had a pronounced leftward shift in the dose/response curves for both integrin activation and P-selectin exposure in the absence of GRK6, but no change in the maximal response. There was also an increase in integrin activation when GRK6−/− platelets were stimulated with ADP, but no increase in P-selectin exposure in response to ADP under these conditions. In addition, there was no increase in either integrin activation or P-selectin exposure in response to the TxA2 analog, U46619, or to convulxin, which is a ligand for the platelet collagen receptor, glycoprotein VI. Platelet aggregation studies (Figure 3B) also showed an increased response to AYPGKF and ADP in GRK6−/− platelets, but no increase in the response to U46619 or convulxin (Figure 3B; supplemental Figure 6).
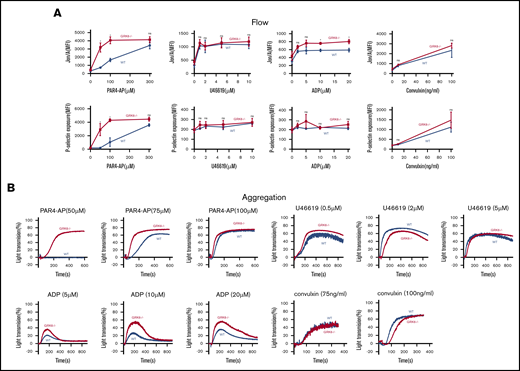
Increased integrin activation, α-granule exocytosis, and aggregation in platelets from GRK6−/−mice. (A) Platelets from GRK6−/− and littermate control mice (WT) were stained with fluorophore-conjugated antibodies to either activated αIIbβ3 (Jon/A antibody) or P-selectin and measured by flow cytometry. Platelets were stimulated with PAR4-AP, ADP, TxA2 mimetic (U46619), or convulxin (CVX) at the concentrations indicated (N = 4). (B) Platelets aggregation in response to a PAR4-AP, ADP, U46619, or CVX at the concentrations indicated (N = 4). *Significant difference (P ≤ .05). MFI, mean fluorescence intensity; ns, not significant.
Increased integrin activation, α-granule exocytosis, and aggregation in platelets from GRK6−/−mice. (A) Platelets from GRK6−/− and littermate control mice (WT) were stained with fluorophore-conjugated antibodies to either activated αIIbβ3 (Jon/A antibody) or P-selectin and measured by flow cytometry. Platelets were stimulated with PAR4-AP, ADP, TxA2 mimetic (U46619), or convulxin (CVX) at the concentrations indicated (N = 4). (B) Platelets aggregation in response to a PAR4-AP, ADP, U46619, or CVX at the concentrations indicated (N = 4). *Significant difference (P ≤ .05). MFI, mean fluorescence intensity; ns, not significant.
Deletion of GRK6 in mice enhances GPCR-dependent signaling in platelets
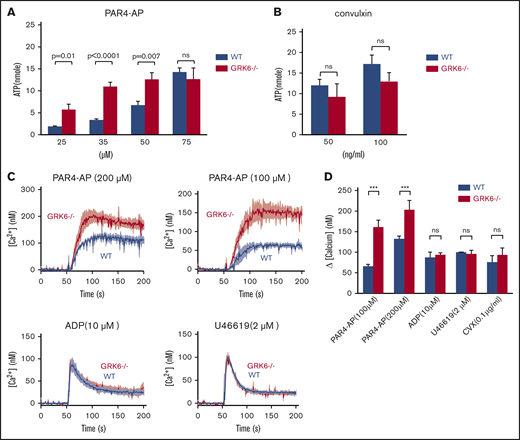
To assess the functional consequences of GRK6 deletion on dense granule secretion, we measured ATP release in platelets from WT and GRK6−/− littermates. ATP and ADP in platelet-dense granules are present in approximately the same equimolar concentrations.22 Single endpoint luminescence assays were used to quantify agonist-induced platelet ATP release.23 There was a similar left shift in the PAR4–AP-mediated dose response for ATP release, but no change in the maximal release (Figure 4A). However, there was no difference in ATP release between WT and GRK6−/− platelets in response to convulxin, indicating that dense granule contents are normal in GRK6−/− platelets (Figure 4B).
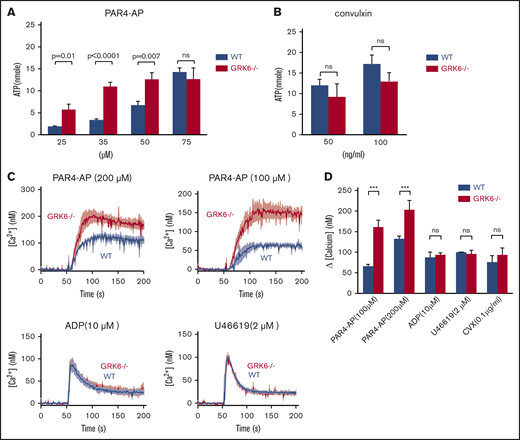
Increased ATP release and Ca2+mobilization in platelets from GRK6−/−mice. (A) ATP release for platelets stimulated with AYPGKF, or (B) CVX at the concentrations indicated (N = 3). (C) Isolated mouse platelets were suspended in Tyrode buffer without Ca2+ and loaded with fura-2 AM (10 μM). The platelets were then washed and resuspended in Tyrode buffer with no extracellular Ca2+ and 0.5 μM EGTA. Changes in fura-2 AM fluorescence were detected with a JASCO FP-8300 spectrofluorometer, exciting at 340 and 380 nm, and measuring emission at 510 nm. Platelets were stimulated with AYPGKF, ADP, U46619, or CVX at the concentrations indicated in the absence of extracellular Ca2+. Representative measurements are shown. (D) The results of 3 experiments (mean ± SEM) are summarized. ***Significant difference (P ≤ .05).
Increased ATP release and Ca2+mobilization in platelets from GRK6−/−mice. (A) ATP release for platelets stimulated with AYPGKF, or (B) CVX at the concentrations indicated (N = 3). (C) Isolated mouse platelets were suspended in Tyrode buffer without Ca2+ and loaded with fura-2 AM (10 μM). The platelets were then washed and resuspended in Tyrode buffer with no extracellular Ca2+ and 0.5 μM EGTA. Changes in fura-2 AM fluorescence were detected with a JASCO FP-8300 spectrofluorometer, exciting at 340 and 380 nm, and measuring emission at 510 nm. Platelets were stimulated with AYPGKF, ADP, U46619, or CVX at the concentrations indicated in the absence of extracellular Ca2+. Representative measurements are shown. (D) The results of 3 experiments (mean ± SEM) are summarized. ***Significant difference (P ≤ .05).
Agonists of which receptors are coupled to Gq cause activation of phospholipase C-β leading to the hydrolysis of phosphotidylinositol-4,5-bisphosphate and the production of diacylglycerol and inositol-1,4,5-trisphosphate. The subsequent increase in [Ca2+]i is due to the release of Ca2+ from intracellular stores followed by the influx of Ca2+ from the plasma membrane, known as store-operated calcium entry. To determine whether deletion of GRK6 affects phospholipase C-β activation and subsequent Ca2+ mobilization, we measured changes in Ca2+ in response to each of the GPCR agonists, with either the fluorescent Ca2+ indicator fura-2 AM, a ratiometric calcium indicator to measure intracellular Ca2+ concentrations, or fluo-4 AM, a nonratiometric Ca2+ indicator to measure influx and efflux that serves in this context to confirm fura-2 AM data. Consistent results were obtained from both assays (Figure 4C-D; supplemental Figure 2). Similar to what we observed in flow cytometric analysis and aggregation assays, PAR4-AP–induced Ca2+ flux showed a higher response in GRK6−/− mice than in WT mice, but the same was not observed for U46619 and convulxin (Figure 4D). In contrast, there was no difference in the Ca2+ response to ADP. Note that Ca2+ mobilization in response to ADP is a readout for P2Y1- and not for P2Y12-mediated signaling. These results suggest that GRK6 targets PAR4, but not P2Y1, thromboxane receptors, or glycoprotein VI in mouse platelets.
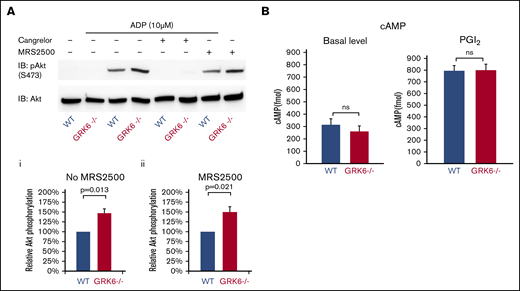
ADP P2Y12 receptors are responsible for the Gi-mediated activation of the serine/threonine kinases, Akt1 and Akt2.24,25 Figure 5A compares ADP-induced Akt activation in WT and GRK6−/− platelets in the presence or absence of cangrelor (a P2Y12 antagonist) or MRS2500 (a P2Y1 antagonist), using Ser473 phosphorylation of Akt as a marker. Resting platelets exhibited negligible Akt phosphorylation whether or not GRK6 was present, but adding ADP caused an increase in Akt phosphorylation that was greater in GRK6−/− platelets than WT controls (Figure 5Aii). Phosphorylation was blocked by cangrelor, but not MRS2500 (Figure 5Aiii), indicating that in both genotypes it was mediated by P2Y12, but not P2Y1.
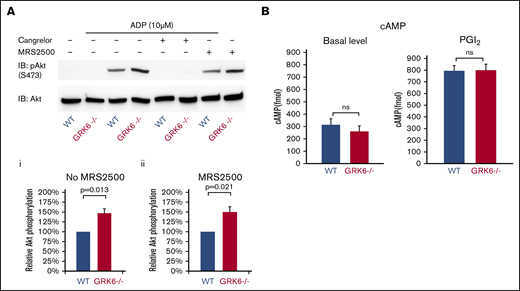
The impact of deleting GRK6 on Akt phosphorylation. (A) Gel-filtered platelets from GRK6−/− mice or matched WT controls were lysed directly or incubated with 10 μM ADP for 5 minutes in the presence or absence of the P2Y1 antagonist MRS2500 (50 μM) or the P2Y12 antagonist cangrelor (100 nM), as indicated. (Ai) Lysates were probed with anti–p-Akt (S473) and reprobed with anti-Akt antibody. (Aii) The p-Akt signal was normalized to the Akt loading control and is represented as signal relative to that of the WT without antagonist and (Aiii) relative to that of the WT under the same conditions in the presence of the P2Y1 antagonist MRS2500. Data are mean ± SEM (N = 3). (B). There is no difference of cAMP formation in WT vs GRK6−/−. (Left) Basal cAMP concentration in GRK6−/− platelets (N = 4) and matched controls (N = 4). (Right) cAMP levels in platelets incubated with PGI2 at the final concentrations 15 μM (mean ± SEM, N = 4). Basal cAMP levels were normal in GRK6−/− platelets, and there was no difference in PGI2-stimulated cAMP formation in WT vs GRK6−/− platelets.
The impact of deleting GRK6 on Akt phosphorylation. (A) Gel-filtered platelets from GRK6−/− mice or matched WT controls were lysed directly or incubated with 10 μM ADP for 5 minutes in the presence or absence of the P2Y1 antagonist MRS2500 (50 μM) or the P2Y12 antagonist cangrelor (100 nM), as indicated. (Ai) Lysates were probed with anti–p-Akt (S473) and reprobed with anti-Akt antibody. (Aii) The p-Akt signal was normalized to the Akt loading control and is represented as signal relative to that of the WT without antagonist and (Aiii) relative to that of the WT under the same conditions in the presence of the P2Y1 antagonist MRS2500. Data are mean ± SEM (N = 3). (B). There is no difference of cAMP formation in WT vs GRK6−/−. (Left) Basal cAMP concentration in GRK6−/− platelets (N = 4) and matched controls (N = 4). (Right) cAMP levels in platelets incubated with PGI2 at the final concentrations 15 μM (mean ± SEM, N = 4). Basal cAMP levels were normal in GRK6−/− platelets, and there was no difference in PGI2-stimulated cAMP formation in WT vs GRK6−/− platelets.
Deletion of GRK6 does not affect PGI2-mediated inhibitory signaling pathway in platelets
Endothelium-derived prostacyclin (PGI2) prevents premature platelet activation and accumulation, and the signaling pathways of PGI2 result in an increase in cyclic adenosine monophosphate (cAMP). It has been suggested that the prostacyclin receptor (IP) is desensitized in platelets.26 We therefore examined the effects of the GRK6 knockout on cAMP levels in resting platelets and in platelets stimulated with PGI2. Our data show that basal levels were normal in GRK6−/− platelets, and there was no difference in PGI2-stimulated cAMP formation in WT vs GRK6−/− platelets (Figure 5B), suggesting that GRK6 is not involved in the inhibitory signaling pathway, which is mediated by endothelial PGI2.
Deletion of GRK6 increases Ca2+ response in MEG-01 cells
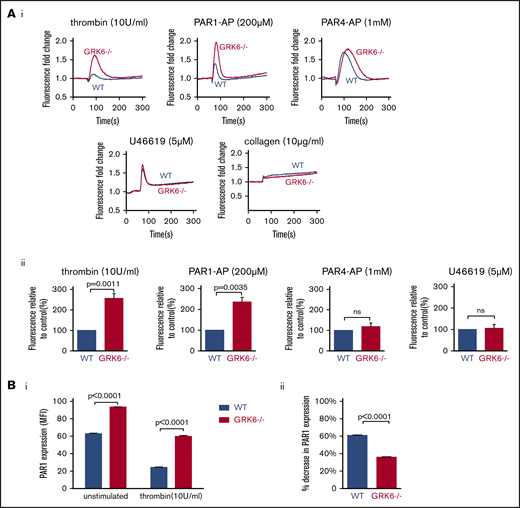
Megakaryoblastic cells, MEG-01, express thrombin receptor PAR1 and PAR4 and have been used as a model to study the signaling of megakaryocytes and platelets.27 In order to understand the function of GRK6 in human megakaryocytes, we generated GRK6 knock-out MEG-01 cell clones using CRISPR-Cas9 techniques with 2 gRNAs targeting at human GRK6 exon 3. Deletion of GRK6 in MEG-01 cells did not affect GRK2 or GRK5 protein expression (supplemental Figure 4; Figure 7Ciii). Deletion of GRK6 did not affect PAR1 protein expression (supplemental Figure 4B). Agonist-induced Ca2+ release was compared between WT and GRK6−/− MEG-01 using fluo-4 AM. Thrombin and the PAR1-AP, SFLLRN, evoked a rapid and significant increase in Ca2+ response in GRK6−/− MEG-01 relative to WT cells. However, there was no difference in Ca2+ response in PAR4-AP stimulated cells (Figure 6A). This indicates that GRK6 is involved in human PAR1 receptor signaling but not in human PAR4 receptor signaling. Consistent with the observation in mouse platelets, there was no difference in U46619- or collagen-induced calcium response between WT control and GRK6−/−. ADP did not cause any response in both WT and GRK6−/− cells (supplemental Figure 3). To investigate the impact of GRK6 deletion on PAR1 surface expression, we used an anti-PAR1 antibody, ATAP2, which recognizes cleaved as well as intact PAR1 receptors.28 Antibody binding to the platelet surface receptors was detected by flow cytometry. Under normal culture conditions, PAR1 expression on the surface of GRK6−/− MEG-01 cells was increased compared with WT cells. Stimulation with thrombin caused a decrease in PAR1 surface expression in both WT and GRK6−/− MEG-01 cells (Figure 6Bi), but this decrease was more pronounced in WT MEG-01 cells than in GRK6−/− MEG-01 cells (Figure 6Bii).
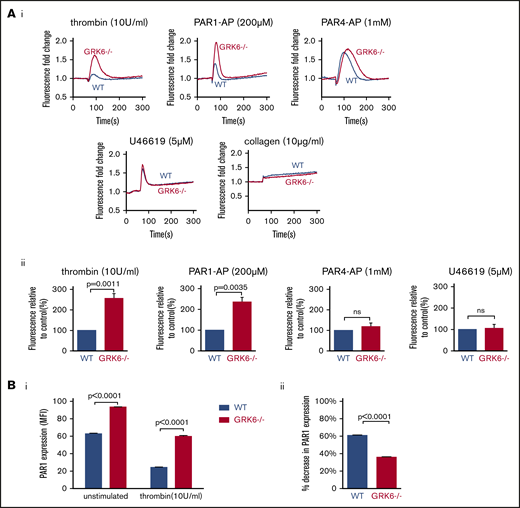
The impact of deleting GRK6 on Ca2+release in MEG-01 cells. (A) MEG-01 cells were loaded with fluo-4 AM, and the cells were stimulated with thrombin (n = 6), PAR1-AP (n = 5), PAR4-AP (n = 8), U46619 (n = 5), or collagen (n = 3), and changes of Ca2+ were recorded. (Ai) The representative trace of Ca2+ response in response to agonists. (Aii) The bar graph summarizes 3 to 8 experiments, respectively (mean ± SEM). (Bi) PAR1 surface expression in WT vs GRK6−/− MEG-01 cells in unstimulated (left) or in response to thrombin stimulation (right), and (Bii) the percentage decrease in PAR1 surface expression in WT and GRK6−/− MEG-01 cells after stimulation (N = 3). An anti-PAR1 antibody (ATAP2), which recognizes cleaved as well as intact PAR1 receptors, was used to detect PAR1 surface expression by flow cytometry.
The impact of deleting GRK6 on Ca2+release in MEG-01 cells. (A) MEG-01 cells were loaded with fluo-4 AM, and the cells were stimulated with thrombin (n = 6), PAR1-AP (n = 5), PAR4-AP (n = 8), U46619 (n = 5), or collagen (n = 3), and changes of Ca2+ were recorded. (Ai) The representative trace of Ca2+ response in response to agonists. (Aii) The bar graph summarizes 3 to 8 experiments, respectively (mean ± SEM). (Bi) PAR1 surface expression in WT vs GRK6−/− MEG-01 cells in unstimulated (left) or in response to thrombin stimulation (right), and (Bii) the percentage decrease in PAR1 surface expression in WT and GRK6−/− MEG-01 cells after stimulation (N = 3). An anti-PAR1 antibody (ATAP2), which recognizes cleaved as well as intact PAR1 receptors, was used to detect PAR1 surface expression by flow cytometry.
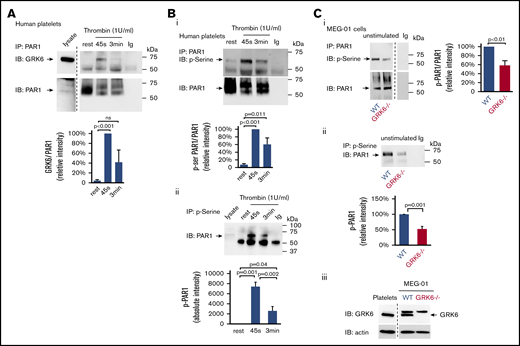
GRK6 forms a complex with PAR1 during platelet activation
It has been reported in nucleated cells that GRKs only transiently interact with GPCRs, and GPCR/GRK interactions are much weaker compared with GPCR/G protein and GPCR/arrestin interactions.29,30 To assess the interaction between GRK6 and PAR1 during platelet activation, resting or activated platelets lysates were subjected to immunoprecipitation. The results show that there is little or no association of GRK6 with PAR1 in resting platelets. Activation of platelets with thrombin caused an increase in binding of GRK6 to PAR1 (Figure 7A). Thus, these results suggest that GRK6 and PAR1 interact directly during platelet activation.
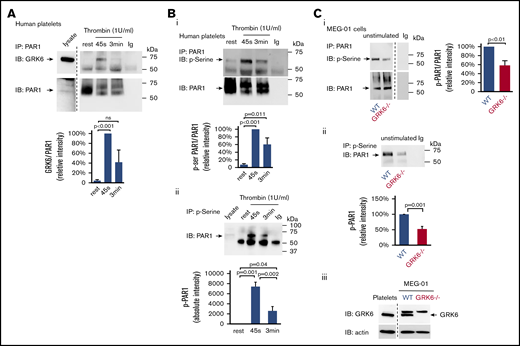
GRK6 forms a complex with PAR1 in activated platelets. (A) Lysates were prepared from resting or thrombin (1 U/mL) activated human platelets. Proteins were precipitated with an anti-PAR1 (ATAP-2) antibody or nonimmune immunoglobulin (Ig) and then probed for GRK6 before reprobing with an anti-PAR1 antibody (N = 5). Note that a marker lane was excised as indicated by the vertical line. (B) Lysates from human platelets incubated with thrombin (1 U/mL) were precipitated with (i) anti-PAR1 (ATAP-2) antibody, then probed with antiphosphoserine antibody before reprobing with an anti-PAR1 antibody, and (ii) antiphosphoserine antibody blot. (Bi) The graph summarizes 6 studies (mean ± SEM). (Bii) The graph summarizes 5 studies (mean ± SEM). (C) Lysates from WT or GRK6−/− MEG-01 were precipitated with anti-PAR1 antibody or nonimmune Ig and then probed with an antiphosphoserine antibody before reprobing with anti-PAR1 antibodies. The graph summarizes 3 studies (mean ± SEM) (Ci). (Cii) Lysates from WT or GRK6−/− MEG-01 were precipitated with antiphosphoserine antibody or nonimmune Ig. The graph summarizes 3 studies (mean ± SEM). (Ciii) Western blot confirmed the loss of GRK6 expression in GRK6−/− clones .
GRK6 forms a complex with PAR1 in activated platelets. (A) Lysates were prepared from resting or thrombin (1 U/mL) activated human platelets. Proteins were precipitated with an anti-PAR1 (ATAP-2) antibody or nonimmune immunoglobulin (Ig) and then probed for GRK6 before reprobing with an anti-PAR1 antibody (N = 5). Note that a marker lane was excised as indicated by the vertical line. (B) Lysates from human platelets incubated with thrombin (1 U/mL) were precipitated with (i) anti-PAR1 (ATAP-2) antibody, then probed with antiphosphoserine antibody before reprobing with an anti-PAR1 antibody, and (ii) antiphosphoserine antibody blot. (Bi) The graph summarizes 6 studies (mean ± SEM). (Bii) The graph summarizes 5 studies (mean ± SEM). (C) Lysates from WT or GRK6−/− MEG-01 were precipitated with anti-PAR1 antibody or nonimmune Ig and then probed with an antiphosphoserine antibody before reprobing with anti-PAR1 antibodies. The graph summarizes 3 studies (mean ± SEM) (Ci). (Cii) Lysates from WT or GRK6−/− MEG-01 were precipitated with antiphosphoserine antibody or nonimmune Ig. The graph summarizes 3 studies (mean ± SEM). (Ciii) Western blot confirmed the loss of GRK6 expression in GRK6−/− clones .
To investigate whether increased binding of GRK6 to PAR1 triggers the phosphorylation of receptors, resting or activated human platelet lysates were immunoprecipitated with an anti-PAR1 antibody and blotted for phosphoserine. Results showed that the level of PAR1 phosphorylation was increased upon thrombin stimulation (Figure 7Bi). Similarly, we detected low levels of PAR1 in immunoprecipitate fractions using antiphosphoserine antibodies in lysates of resting platelets. However, PAR1 was highly enriched in phosphoserine immunoprecipitate fractions following platelet activation (Figure 7Bii), further supporting increased PAR1 serine phosphorylation in response to thrombin activation in platelets. To investigate further the role of GRK6 in the phosphorylation of PAR1, we examined the level of PAR1 phosphorylation in both WT and in GRK6−/− MEG-01 cells. Deletion of GRK6 in MEG-01 cells caused a significant decrease in PAR1 phosphorylation in unstimulated cells (Figure 7Ci-Cii). The remaining phosphorylation of PAR1 in GRK6−/− MEG-01 cells might be due to GRK2 or GRK5 expressed in the cells (supplemental Figure 4Aii-iii). Collectively, these observations suggest that GRK6 is involved in PAR1 phosphorylation during platelet activation.
Discussion
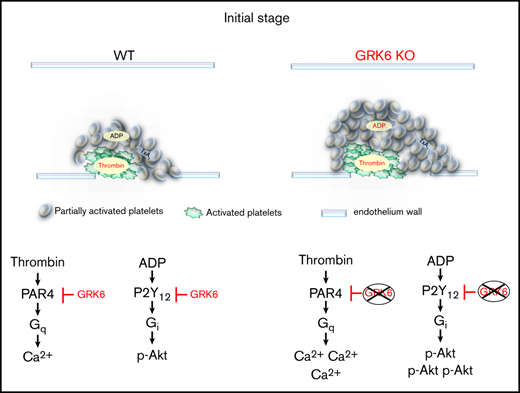
Although initial adhesion of platelets to the vessel wall is driven by collagen, the subsequent recruitment of additional platelets into a growing thrombus requires mediators such as thrombin, TxA2, and ADP, all of which act through GPCRs.31,32 Recently, a model of the hemostatic response has been established in which the development of gradients of platelet-soluble agonists presents within the evolving platelet plug, resulting in a gradient of platelet activation emanating from the injury site.18,19,33-35 In the core region of the thrombus, thrombin mediates platelet activation with minimal requirement of ADP and TxA2. In contrast, ADP and TxA2 signaling are critical for outer shell region formation. It has been long recognized that during platelet activation, GPCRs undergo desensitization following activation. For example, (1) PAR1 undergoes rapid desensitization due to internalization, whereas activation-dependent internalization of PAR4 is much slower than that seen for PAR1.28,36-38 However, whether PAR1 or PAR4 receptor desensitization is regulated by GRKs in platelets remains undetermined. (2) Platelets also become desensitized to activation upon continued exposure to ADP. Hardy et al show, using a transfected cells–based assay, that P2Y12 receptors undergo functional desensitization, and that GRK2 and GRK6 are involved.16,39 However, whether this mode of regulation exists in anucleate platelets is unclear. Furthermore, the function of GRKs during hemostasis and thrombosis is unknown.
In this study we asked whether platelet activation is regulated at the receptor level to limit GPCR signaling. The results show that such a rate-limiting mechanism is present in platelets involving GRK6. GRK6 functions as a primary checkpoint to limit the intensity and duration of signaling via particular GPCRs during platelet activation. Deleting GRK6 in mouse platelets increases platelet reactivity in a PAR4- and P2Y12-dependent manner. Total platelet accumulation is also increased in GRK6−/− mice, which is primarily due to an accelerated rate of early-phase platelet accumulation. Next, we investigated the signaling mechanisms that might underlie the observed phenotypic change. We demonstrate that cytosolic Ca2+ release and α-granule secretion are increased when platelets are activated with PAR4-AP. Akt activation is also increased in GRK6−/− platelets in response to ADP stimulation. Collectively, our observations show for the first time that deletion of GRK6 produces a pathway-selective gain of function in platelets (supplemental Figure 5). The gain of function is confined to both PAR4-mediated signaling, leading to elevated Ca2+ mobilization and ATP release, and P2Y12-mediated signaling, resulting in increased phosphorylation of Akt.
Mouse platelets express PAR3 and PAR4. PAR4 is the principal thrombin receptor in mouse platelets. Thrombin signaling in mouse platelets is mediated by PAR3-faciliated cleavage of PAR4. As noted previously, thrombin is generated close to the site of injury and can induce maximal platelet activation independent of TxA2 and P2Y12 signaling.18,35 This is consistent with our observation that the initial hemostatic plug forms much faster in GRK6−/− mice than in WT control, with an increase in the rate of the platelet accumulation. Furthermore, the increased size of the core region is driven by the gradient of thrombin concentration emanating from the site of the injury. Consistent with the leftward shift in the dose/response curve of GRK6−/− platelets to the PAR4 agonist stimulation that we observed, thrombin-driven platelet activation and corresponding deposition in GRK6−/− mice continue to occur further out from the injury site, where the thrombin concentration is too low to trigger a response in WT platelets. During plug extension, additional platelet recruitment in the outer shell region requires ADP and TxA2. In this study, among the agonists tested, PAR4 peptide produced the greatest responses in flow, α-granule exocytosis, and Ca2+ mobilization assays. ADP stimulation also generated a significant increase in integrin activation, platelet aggregation, and P2Y12-mediated Akt activation, but had no effect on Ca2+ mobilization, which is P2Y1 mediated. Notably, knocking out GRK6 does not affect TxA2-mediated signaling. TxA2 signals through the thromboxane receptor (TP) receptor, and the TP receptor is encoded by a single gene that can be alternatively spliced in the carboxyl-terminal tail leading to 2 variants, TPα (343 residues) and TPβ (407 residues), that share the first 328 amino acids. In nucleated cells, it has been shown that the TPβ, but not the TPα, undergoes agonist-induced internalization.40 TxA2-stimulated effects in platelets are mediated predominantly through the α isoform.41 Because GRK6 mainly phosphorylates the C-terminus of GPCRs, we propose that GRK6 cannot phosphorylate TPα and thus does not regulate platelet TxA2 signaling because TPα has shorter C-terminus. In addition, our data also suggest that GRK6 is not involved in the inhibitory signaling pathway mediated by endothelial PGI2.
In human platelets, thrombin signals through 2 GPCRs, PAR1 and PAR4. In contrast to mouse PAR4 and PAR3, human PAR1 and PAR4 have functional differences that are complementary, and both are essential for platelet activation, although at a different scale and with different signaling kinetics.42,43 Our current studies in human MEG-01 cells show that there is an increase in Ca2+ response to PAR1 agonist when GRK6 is absent, but no change in Ca2+ response to PAR4 agonist. Why is there an effect on mouse PAR4 but not human PAR4? First, human PAR4 has been shown to internalize much less robustly than PAR1, likely because human PAR4 has fewer Ser/Thr phosphorylation sites than human PAR1.37 Furthermore, the C-terminus of human PAR4 has fewer Ser/Thr phosphorylation sites compared with mouse PAR4. Thus, it would be interesting to compare putative GRK6-mediated phosphorylation sites in human PAR4 vs mouse PAR4 in a future study. The deletion of GRK6 does not affect TxA2-induced Ca2+ response, which mimics what we have seen in GRK6−/− mouse platelets. MEG-01 cells normally do not respond well to weaker agonists, such as collagen and ADP,44 and as such we cannot at this point compare the response of MEG-01 cells to these agonists with what we have seen in mouse platelets. Finally, the results from our current study show that platelet activation by thrombin causes increased association between GRK6 and PAR1, indicating that GRK6 is involved in PAR1-mediated signaling in platelets. Consistent with increased GRK6 binding to PAR1, there is an increase in serine phosphorylation of PAR1 in human platelets in response to thrombin stimulation. Along with this, we observed a decrease in PAR1 internalization in MEG-01 cells lacking GRK6, and deletion of GRK6 in MEG-01 cells caused a decrease in serine phosphorylation of PAR1. Collectively, this places GRK6 at a checkpoint at the receptor level that balances the need for platelets to respond quickly in response to vascular injury while retaining its function as a molecular brake acting on PAR signaling, limiting platelet activity.
Along with GRK6, there are several other members of the GRK family expressed in platelets.14-16 The gain-of-function phenotype in GRK6−/− mice suggests that GRK6, GRK5, and GRK2 may not be fully redundant. This conclusion awaits studies on platelets lacking GRK5 or GRK2. Finally, in nucleated cells, GRKs are generally considered to regulate receptor desensitization via their canonical role (GRK-mediated GPCR phosphorylation), and GRKs-mediated GPCR desensitization can occur rapidly in some cell types. However, GRK2 and GRK5 can also mediate GPCR signaling through noncanonical actions (kinase-independent molecular interactions). Interestingly, GRK6−/− mice show an increased rate of initial platelet accumulation in vivo in response to injury, suggesting that the effect of GRK6 deletion is primarily on the early stages of platelet activation. GPCR desensitization in platelets, a process that may serve as a protective mechanism to prevent excessive platelet accumulation, has generally been thought of as a late signaling event in platelets. However, this statement has not been experimentally validated in vivo. Interestingly, desensitization of epinephrine-initiated human platelet aggregation occurs rapidly, with a t1/2 of 3 minutes for desensitization.45 Our studies show that the maximum difference between platelet accumulation in GRK6−/− and WT mice is most prominent at early time points. This indicates that GRK6-mediated GPCR desensitization actually occurs as an early signaling event during platelet activation. Characterizing the underlying mechanism by which GRK6 regulates the receptor-level signaling node, via either canonical (kinase-dependent mechanism) or noncanonical actions (kinase-independent mechanism) during platelet activation, is worthy to investigate in a future study.
In summary, the present studies demonstrate that platelet activation downstream of GPCR-mediated signaling is limited by GRK6 at the level of receptors. This leads us to suggest that the existence of regulatory mechanism at the receptor level allows for tight regulation of platelet activation even at its earliest stages. Combined with what is known about the function of RGS proteins in platelets, further investigating how GPCR and G protein signaling can be regulated during platelet activation would provide a fruitful avenue for understanding the hemostatic response to vascular injuries.
Acknowledgments
This work was supported by American Heart Association grant 14SDG20380473 and National Institutes of Health, National Heart, Lung, and Blood Institute grants R01HL144574 (P.M.), P01 HL40387 (L.F.B.), R01HL137207 (L.E.G.), and R01HL142959 (U.P.N.).
Authorship
Contribution: X.C., S.G., M.C., D.D., J.G.T.W., X.Z., M.U.N., and P.M. performed the experiments; X.C., S.G., T.J.S., L.F.B., J.B., L.E.G., S.E.M., U.P.N., and P.M. designed the experiments; and X.C. and P.M. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Peisong Ma, Department of Medicine, Sidney Kimmel Medical College, Thomas Jefferson University, 1020 Locust St, Room 394, Philadelphia, PA 19107; e-mail: peisong.ma@jefferson.edu.

![Increased platelet accumulation in response to hemostatic injury. (A) Representative images of hemostatic plugs formed in WT and GRK6−/− mice 1 minute after injury (Ai). (Aii) Confocal intravital fluorescence microscopy was performed to follow platelet accumulation and P-selectin expression. Data are mean ± SEM; 46 injuries in 5 WT mice and 42 injuries in 5 GRK6−/− mice. (Aiii) Graph shows the area under CD41 vs time curve (area under the curve [AUC]). (Aiv) Graph shows the area under P-selectin (+) vs time curve (AUC). The line and error bars show the median and interquartile range. (B) CD41+ area at 45 seconds after injury (Bi). (Bii) The rate of CD41 deposition at 45 seconds after injury, and the bar graph represents the average slope of the liner trend line equation of each injury. Forty-one injuries in 5 WT mice and 41 injuries in 5 GRK6−/− mice. (C) Fibrin deposition after making small penetrating injuries in the cremaster muscle arterioles with laser in GRK6−/− mice and littermate controls. Data are mean ± SEM; 24 injuries in 5 WT mice and 25 injuries in 5 GRK6−/− mice (Ci). (Cii) Bar graphs represent the fibrin accumulation at the end of the 3-minute observation period.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/1/10.1182_bloodadvances.2019000467/3/m_advancesadv2019000467f2.png?Expires=1768444100&Signature=mboznnr0vCH5DXzrxht8Zo0bnDcNqorKOalx7UVvZqE7HeiLJCcdtgs-YGGrXOlIFHgsxM6TLZFR3r0~-fwQXe8Rspcyw1hWPy04SNr3FFUOdUagEmodz-O2rhhEQGghaID8qsxhSuKVzOqrUtpJgGNtinPWEUs7Q3eWsktRNQ97hZ2YVzGhrDZbQXGBw~wyMsDmxsuYeirWNG5aq4JN4u6jmSCOG1XuE0npbkSnbBzf7XnFys3~O8514x-1G3mgI~wilAs9QpsqUMjuw9-nsXPa1mV19QdA0Rjx8uNHS72i2WdNngSW3vlMEbciPQ6-o1oeO5Ogu5UZTQHsxXCEdQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)