Key Points
Active comorbidity, second transplant, and aGVHD grades III to IV are significant risk factors for TA-TMA.
TA-TMA might represent a form of vascular GVHD, and therefore continuing control of aGVHD is important to prevent worsening of TA-TMA.

Abstract
This study aimed to identify a risk profile for development of transplant-associated thrombotic microangiopathy (TA-TMA) in children undergoing hematopoietic stem cell transplantation (HSCT). Between 2013 and 2016, 439 children underwent 474 HSCTs at 2 supraregional United Kingdom centers. At a median of 153 days post-HSCT, TA-TMA occurred among 25 of 441 evaluable cases (5.6%) with no evidence of center variation. Sex, underlying disease, intensity of the conditioning, total body irradiation–based conditioning, the use of calcineurin inhibitors, venoocclusive disease, and viral reactivation did not influence the development of TA-TMA. Donor type: matched sibling donor/matched family donor vs matched unrelated donor vs mismatched unrelated donor/haplo-HSCT, showed a trend toward the development of TA-TMA in 1.8% vs 6.1% vs 8.3%, respectively. Presence of active comorbidity was associated with an increased risk for TA-TMA; 13% vs 3.7% in the absence of comorbidity. The risk of TA-TMA was threefold higher among patients who received >1 transplant. TA-TMA rates were significantly higher among patients with acute graft-versus-host disease (aGVHD) grades III to IV vs aGVHD grade 0 to II. On multivariate analysis, the presence of active comorbidity, >1 transplant, aGVHD grade III to IV were risk factors for TA-TMA (odds ratio [OR]: 5.1, 5.2, and 26.9; respectively), whereas the use of cyclosporine A/tacrolimus-based GVHD prophylaxis was not a risk factor for TA-TMA (OR: 0.3). Active comorbidity, subsequent transplant, and aGVHD grades III to IV were significant risk factors for TA-TMA. TA-TMA might represent a form of a vascular GVHD, and therefore, continuing control of aGVHD is important to prevent worsening of TA-TMA associated with GVHD.
Introduction
Transplant-associated thrombotic microangiopathy (TA-TMA) represents a diagnostic challenge for clinicians and can lead to multiorgan failure and death.1,2 There is a wide variation in the prevalence of TA-TMA across different reports ranging between 0.5% and 76%3-8 with a mortality rate reaching as high as 80%.7,9,10 This variation probably reflects the lack of consistency in the diagnostic criteria for TA-TMA. A previous review in 2004 pointed to the presence of 38 different diagnostic criteria being used in 35 studies, including 447 patients with TA-TMA.6 Moreover, the pathophysiology of TA-TMA is poorly understood, and histopathological features overlap with other small vessel injury diseases.11 Recent studies have focused on genetic predisposition and abnormal activation of the complement cascade to achieve a more refined definition of this condition.12-15 Different risk factors have been associated with a higher risk of developing TA-TMA16 : calcineurin inhibitors (eg, tacrolimus) and sirolimus have been blamed for increasing the risk of endothelial damage,7,17,18 although the beneficial effect of early discontinuation of these agents remains to be proven16 and must be balanced with the risk of inducing or worsening graft-versus-host disease (GVHD). Screening of patients for risk factors of TA-TMA before transplant has been problematic, and close monitoring for early signs of microangiopathy is necessary to recognize this complication before severe manifestations occur1 since early intervention can provide a better outcome.19,20 Recently reviewed criteria for diagnosis of TA-TMA have included clinical and laboratory findings1 and should help clinicians to identify patients with early-stage TA-TMA and those at a higher risk of a poor outcome. Recognition of pretransplant variables that could identify high-risk patients might permit prophylaxis or even tailoring of the transplant procedure (eg, conditioning, GVHD prophylaxis). Risk factors have been variably reported in adults,21 and data on children remain scarce. We aimed to identify a risk profile for the development of TA-TMA in children undergoing hematopoietic stem cell transplantation (HSCT) and to explore potential correlation with other posttransplant complications that could improve our comprehension of TA-TMA.
Methods
Patients characteristics
Records of patients who underwent allogeneic HSCT at the 2 supraregional UK centers, Great Ormond Street Hospital (GOSH) for Children, London and The Great North Children’s Hospital (GNCH), Newcastle upon Tyne, between January 2013 and December 2017 were analyzed.
TA-TMA was defined based on the criteria established by Jodele et al, 2016.1 The diagnostic criteria included either a biopsy-proven diagnosis or the presence of 5 out of 7 laboratory criteria. Biopsy-proven diagnosis of TA-TMA relied on histologic evidence of microangiopathy as described by Jodele et al, 2015.9 This includes the presence of denuded and injured endothelium, microthrombosis, and schistocyte extravasation into the interstitium. Biopsy slides were reviewed and reported by experienced histopathologists in both BMT centers who could differentiate TA-TMA from GVHD. Informed consent was obtained from the parents of all children.
Within the pediatric population, 88% to 92% of patients develop TA-TMA before day 100 post-HSCT.21,22 Hence, patients who developed TA-TMA at any time posttransplant and patients who survived the first 100 days posttransplant or had a follow-up of at least 100 days posttransplant and prior any second intervention (if needed) were considered evaluable for the study. Risk factors for TA-TMA were divided into the following: patient-derived factors, transplant-related factors, and posttransplant (complication)-related factors.
Patient-derived factors included sex, age at transplant, primary diagnosis, and presence of active comorbidity at day 0 (D0). Active comorbidity was defined as factors that can induce endothelial dysfunction. They included uncontrolled bacterial, fungal, or viral infection requiring continuation of antimicrobial treatment past D0 of transplant, cardiovascular disease requiring specific therapy, respiratory compromise requiring oxygen therapy or assisted ventilation, or the use of prednisolone therapy ≥0.3 mg/kg for control of autoimmune disease or active inflammation induced by GVHD (from prior transplant), Omenn syndrome, or active disease.
Transplant-related factors included intensity of preparative conditioning regimen, the use of total body irradiation (TBI)–based conditioning, the use of serotherapy, stem cell source, stem cell dose, degree of recipient-donor HLA match, and donor source. Posttransplant (complication)-related factors included the development of posttransplant viral reactivation in the first 100 days of transplant, development of venoocclusive disease (VOD), or acute graft-versus-host disease (aGVHD).
Stem cell source, donor source, HLA typing, and preparative conditioning protocol
Bone marrow (BM), peripheral blood stem cells (PBSCs), and cord blood were used as stem cell sources. High-resolution typing was performed by molecular typing (at allelic level) for HLA-A, -B-C, -DR, and -DQ loci. Donors were divided into matched donors (10 out of 10 HLA match); matched sibling donor (MSD), matched family donor (MFD), and matched unrelated donor, mismatched related family or mismatched unrelated donor (MMFD, MMUD), and haploidentical donors (5 to 6/10 matched). Mismatched donors included 8 to 9 out of 10 HLA match for all stem cell sources and including 7 out of 10 HLA match for recipients of cord blood.
Preparative regimens were defined as the following: reduced intensity conditioning (RIC) protocols including treosulfan/fludarabine (Treo/Flu) or fludarabine/melphalan (Flu/Mel) or RIC busulfan/fludarabine (Bu/Flu) targeting Bu area under the curve 45 to 65 mg*h/L. Myeloablative protocols (myeloablative conditioning [MAC]) included myeloablative Bu (targeted Bu area under the curve >70 mg*h/L) in conjunction with cyclophosphamide (Cyc) or Flu ± Mel, Treo/Flu/thiotepa (Treo/Flu/TT), TBI (12.2 to 14.4 Gy)/Cyc based conditioning, or the use of BEAM (carmustine, etoposide, cytarabine, melphalan). Minimal intensity conditioning included Flu/Cyc, or low-dose TBI (3 Gy)/Flu or TT (5 mg/kg). In vivo T-cell depletion involved the use of either rabbit antithymocyte globulin or alemtuzumab. Ex vivo T-cell depletion involved the use of TCRαβ/CD19 depletion for haplo-HSCT or mismatched grafts.
Supportive care
All patients were nursed in single rooms with laminar flow. Supportive therapy included antimicrobial prophylaxis as per institutional practice (cotrimoxazole prophylaxis was given in both centers in addition to ciprofloxacin in London). Cotrimoxazole was given throughout the transplant in Newcastle, whereas cotrimoxazole was discontinued on D−1 in London to be restarted once absolute neutrophil counts were ≥1000 cells/μL (usually around D+28). In both centers, cotrimoxazole was completely stopped once the patient was off cyclosporine and had a CD4 count >300 cells/μL. In London, ciprofloxacin in a dose of 10 mg/kg was given twice daily until absolute neutrophil counts were ≥1000 cells/μL. Based on primary diagnosis, patients received immunoglobulin replacement until B-cell function recovery and ursodeoxycholic acid until D+28. All patients received acyclovir prophylaxis that was discontinued once the patient was off cyclosporine with a CD4 ≥300 cells/μL (until at least 1 year post-HSCT).
Posttransplant viral reactivation
The presence of virus detected by polymerase chain reaction in blood (cytomegalovirus [CMV], Epstein-Barr virus [EBV], adenovirus) in both centers was recorded weekly from D−10 onwards.
VOD
The diagnosis of VOD was based on clinical criteria according to the modified Seattle criteria.23
GVHD
Grading of aGVHD was performed according to Seattle criteria.24
Relapse
Relapse was defined as reappearance of blasts in peripheral blood, recurrence of BM blasts of >5%, or extramedullary disease infiltrates at any sites.
Statistical analysis
Statistical analysis was performed using SPSS, version 24. Descriptive analyses were performed using median, mean, minimum, and maximum. Parametric data were analyzed using 1-way analysis of variance and post hoc test. Transplant-related mortality (TRM) was analyzed using Kaplan-Meier estimates and log-rank test. A comparison with 2-sided P < .05 was statistically significant. Variables reaching P < .10 in univariate analysis for development of TA-TMA were included in Cox proportional hazard regression models using a backward stepwise selection. The threshold for statistical significance for all tests was set to P < .05.
Results
Patient characteristics
Four hundred thirty-nine children received 474 HSCTs at the 2 centers, GNCH (n = 185) and GOSH (n = 254), during the study period. One hundred forty-seven patients have been previously reported.25-29 Transplants (441/474) were evaluable for the study. For the excluded patients (33/474), the median day to mortality was 15 days. These patients had such a short follow-up that they would contribute very little to the analysis.
Among the 441 grafts, 180 were carried out at GNCH and 261 were carried out at GOSH for management of metabolic (n = 26), hematological condition (n = 148), or primary immune deficiency (PID) (n = 267). A list of diagnoses across different transplants is shown in supplemental Table E1. Twenty-five patients developed TA-TMA at a median of 153 days (range: 21 days to 528 days). There was no center variation in the risk for TA-TMA: 9 out of 180 GNCH (5%) vs 16 out of 261 GOSH (6.1%) (P = .15).
Patient-derived factors
Sex, underlying diagnosis, and age at transplant are not associated with TA-TMA.
Seventeen boys (17/288; 5.9%) and 8 girls (8/153; 5.2%) developed TA-TMA (P = .31). Underlying diagnosis: metabolic disease (0/26); hematological disease (8/148; 5.4%); or PID (17/267; 6.3%), did not influence the risk of TA-TMA (P = 1). The median age at transplant was 2.7 years (range: 0.14 to 15.6 years) in the patients who did not develop TA-TMA vs 2.3 years (0.65 to 13 years) in the group who developed TA-TMA (P = .9). Supplemental Table E2 lists all the factors that were not associated with TA-TMA.
Active comorbidity at D0 is a risk factor for TA-TMA.
Active comorbid condition at D0 was recorded among 93 of 441 of the studied cohort (21%). Uncontrolled infection at D0 was recorded among 41 transplants: fungal infection (n = 29); bacterial infection (n = 2); viral infection (n = 9); and uncontrolled cryptosporidium disease (n = 1). Cardiovascular instability was recorded in 6 transplants in the form of cardiomyopathy requiring antifailure treatment (n = 3) or cardiac thrombus requiring continuation of anticoagulant therapy (n = 3). Continuation of steroid therapy to control active inflammation was recorded among 28 cases: 6 for active Omenn syndrome; 4 for active BCGiosis; 11 for control of autoimmune disease; 2 for the control of active GVHD; 5 for control of active disease (hemophagocytic lymphohistiocytosis [HLH]). Pulmonary instability with poor lung function and requirement of oxygen therapy or assisted ventilation was seen among 18 transplants.
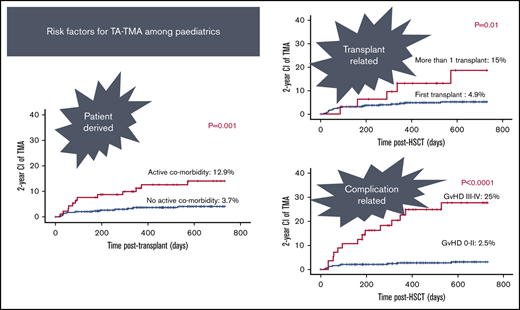
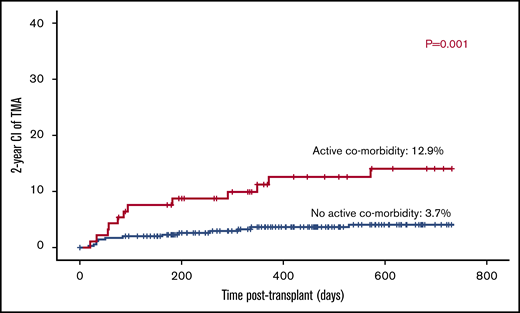
Twelve out of 93 patients with active comorbidity developed TA-TMA vs 13 out of 348 patients with no active comorbid condition (12.9% vs 3.7%; P = .001; Figure 1).
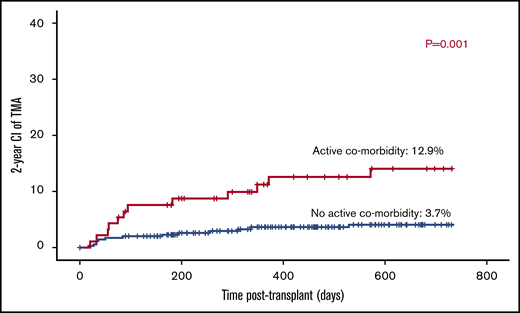
Transplant-related factors
Conditioning intensity and TBI-based conditioning are not risk factors of TA-TMA.
The intensity of conditioning did not influence the development of TA-TMA (P = .69). TA-TMA was reported among 12 out of 227 (5.2%) patients who used RIC/minimal intensity conditioning vs 13 out of 193 (6.7%) patients who received MAC conditioning.
TBI was included in the conditioning regimen of 51 transplants: 44 received full-dose TBI, whereas 7 received low-dose TBI. Three patients with TBI conditioned graft developed TA-TMA (3/51; 5.8%) vs of 22 out of 390 (5.6%) who did not receive TBI (P = 1).
The use of serotherapy was not associated with TA-TMA.
Serotherapy in the form of rabbit antithymocyte globulin (dose: 7.5 to 15 mg/kg; n = 196) or alemtuzumab (0.3 to 1 mg/kg; n = 146) was included in the conditioning regimen of 341 grafts (77%). TA-TMA occurred among 21 (6.1%) out of 342 patients who received serotherapy vs 4 (4%) out of 99 patients whose transplant did not include serotherapy (P = .4).
PBSCs are associated with a slightly higher risk for TA-TMA.
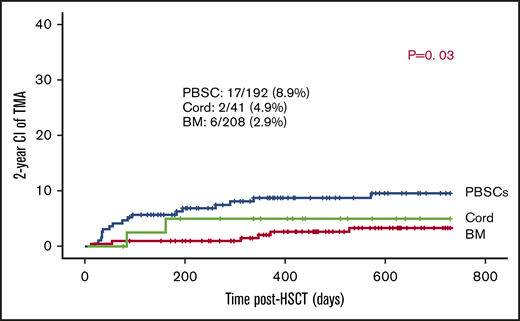
Of the transplants, 90.7% used BM (n = 208) or PBSCs (n = 192) as stem cell sources. In the remaining 41, cord was the stem cell source. The use of PBSCs was associated with slightly higher risk of TA-TMA: 17 out of 192 (8.9%) vs 2 out of 41 (4.9%) of cord grafts and 6 out of 208 of BM grafts (2.9%) (P = .03; Figure 2).
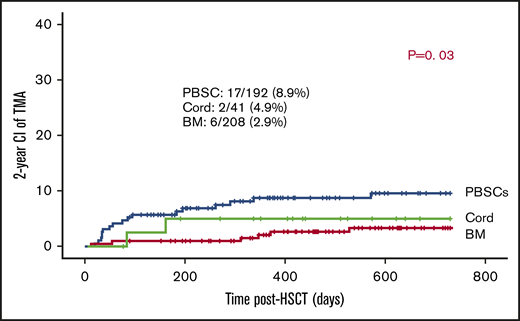
CD34 stem cell dose does not influence the development of TA-TMA.
There was no difference in the median CD34 dose between patients who developed TA-TMA vs patients who did not develop TA-TMA: 10.6 × 106/kg (range: 0.33 to 33.9 × 106/kg) vs 8 × 106/kg (range: 0.07 to 56.6 × 106/kg) (P = .63). One hundred eighty-eight patients received a CD34 dose ≥10 × 106/kg. TA-TMA was slightly higher among (7.9%) recipients of a megadose of D34 (15 out of 188; 7.9%) vs patients who had a stem cell dose <10 million per kilogram (10/253; 3.9%); however, the difference was statistically insignificant (P = .124).
Number of transplants increases the risk for TA-TMA.
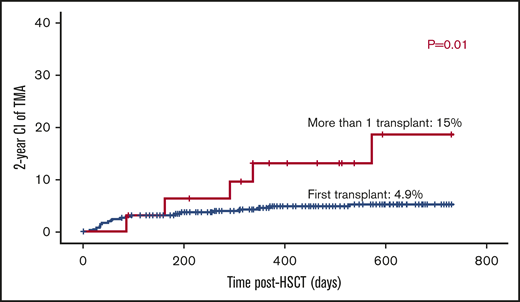
The majority of patients received 1 transplant (n = 409). Thirty-two had >1 transplant; 2 grafts (n = 28), and 3 grafts (n = 4). The risk of TA-TMA was threefold higher among patients who received >1 transplant: 5 out of 32 (15%) vs patients who received only 1 transplant: 20 out of 409 (4.9% P = .01; Figure 3).
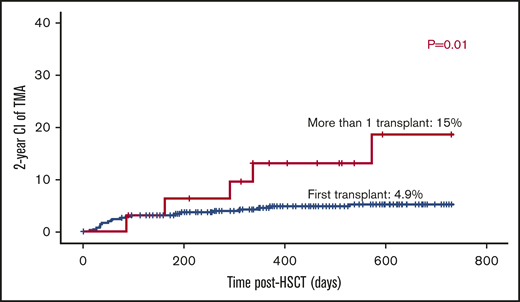
Mismatched grafts have slightly higher risk for TA-TMA.
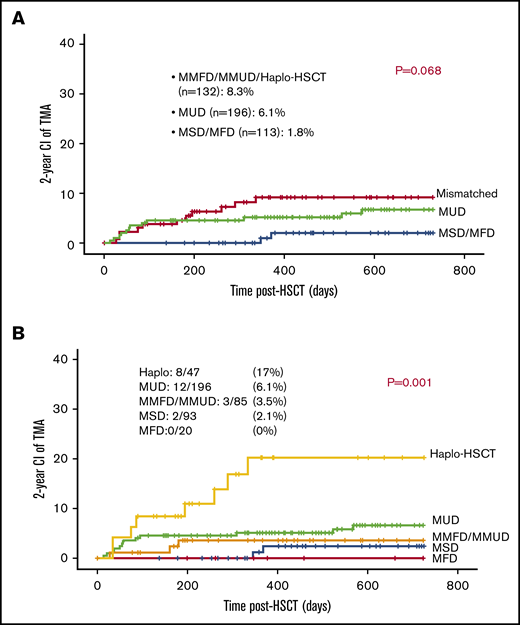
The majority of patients (70%) received 10 out of 10 HLA-matched grafts: either MUD (n = 196), MSD (n = 93), or MFD (n = 20). Eighty-five patients received 1 antigen to 3 antigen mismatched related or unrelated grafts (MMFD, MMUD) and 47 received a haploidentical parental graft. TA-TMA was slightly more frequent among patients who received either mismatched or haploidentical grafts (11/132; 8.3%) vs patients who received MUD graft (12/196; 6.1%) and patients who received either MFD or MSD (2 out of 113; 1.7%; P = .068; Figure 4A).
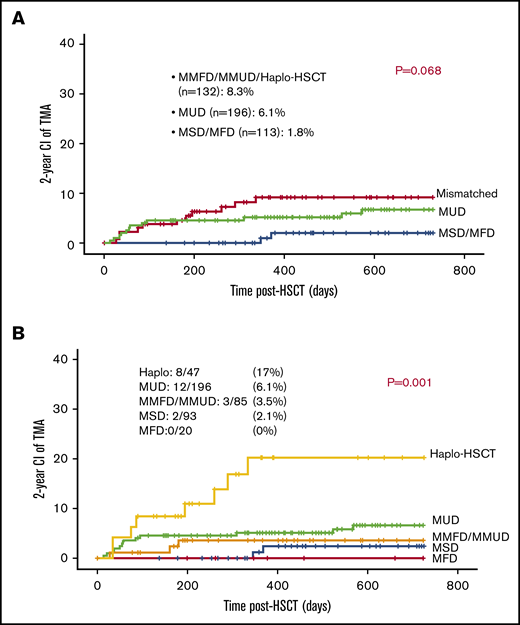
Mismatched grafts and TA-TMA. (A) Mismatched grafts are slightly associated with increased risk for TA-TMA. (B) Haplo-HSCT was associated with increased risk for TA-TMA.
Mismatched grafts and TA-TMA. (A) Mismatched grafts are slightly associated with increased risk for TA-TMA. (B) Haplo-HSCT was associated with increased risk for TA-TMA.
Active comorbidity and high grades of aGVHD are risk factors for TA-TMA among haploidentical grafts.
Recipients of haploidentical grafts had significantly higher risk for TA-TMA approaching 17% (8/47), whereas recipients of other grafts had a risk of 0% to 6% to develop TA-TMA: 12 out of 196 (6.1%) among recipient of MUD grafts; 3 out of 85 (3.5%) among recipients of MMFD or MMUD grafts; 2 out of 93 (2.1%) among recipients of MSD grafts; and 0 out of 20 (0%) among recipients of MFD grafts (P = .001; Figure 4B).
More analysis was carried out among this particular group to reveal possible cofounder factors contributing to the high incidence of TA-TMA among recipients of haploidentical grafts. Ten patients had active comorbid condition at D0: 4 of them (40%) developed TA-TMA, whereas 37 had no active comorbidity at D0 and only 10.8% (4 patients) developed TA-TMA (P = .05). Forty-two patients received only 1 transplant and 5 of them developed TA-TMA (11.9%), whereas 3 out of 5 patients (60%) who had a subsequent graft developed TA-TMA (P = .05). Fifteen patients developed aGVHD grade II to IV: 6 of them developed TA-TMA (40%), whereas 32 had low grades of aGVHD, grade 0 to I, and only 6% developed TA-TMA (2 patients) (P = .009). Of note, 5 patients developed grade III to IV aGVHD, and 4 of them (80%) developed TA-TMA, whereas 42 had aGVHD grade 0 to II and only 4 (9.5%) developed TA-TMA (P = .002). Interestingly, both TBI-based conditioning and the use of calcineurin inhibitors were not associated with TA-TMA among recipients of haploidentical grafts (P = .26 and P = 1; respectively; Table 1).
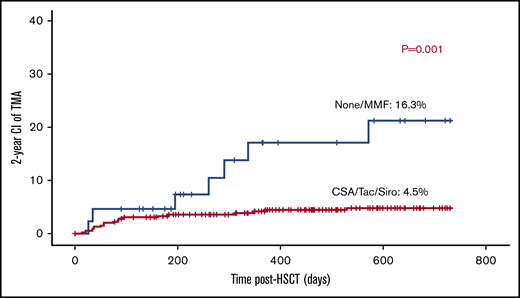
Use of calcineurin inhibitors or sirolimus is not a risk factor for TA-TMA.
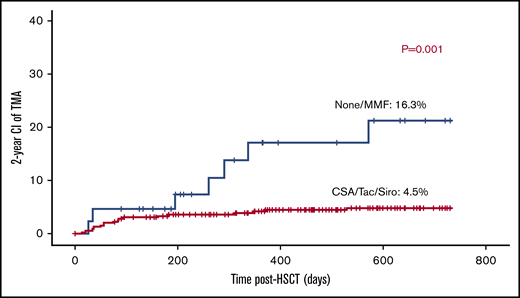
Cyc-based GVHD prophylaxis either alone (n = 70) or in conjunction with mycophenolate mofetil (MMF; n = 275), methotrexate (n = 46), or prednisolone (n = 2) was used in 393 transplants (89%). Tacrolimus-based GVHD prophylaxis and sirolimus-based GVHD prophylaxis were given to 4 and 1 patients; respectively. MMF-only aGVHD prophylaxis was given to 2 patients. Although 41 patients did not receive any GVHD prophylaxis, 29 received a TCRαβ/CD19-depleted graft, 3 had MMUD graft (2 were αβ/CD19 depleted), 6 had MSD, 2 had MFD, and 1 had an MUD graft. Surprisingly, the use of calcineurin inhibitors or sirolimus was associated with 4.7% risk of TA-TMA (19/399) vs 14.2% (6/42) in patients who either received no prophylaxis or only MMF (P = .002; Figure 5).
Posttransplant related factors
Posttransplant viremia is not associated with TA-TMA.
One hundred days of viral reactivation of either CMV viremia, EBV viremia, or AdV viremia were recorded among 200 out of 441 grafts (45.5%). Viral reactivation was not associated with increased risk for TA-TMA. TA-TMA was recorded among 15 out of 200 patients with viral reactivation (7.5%) vs 10 out of 241 patients with no history of viral reactivation (4.1%; P = .68).
Posttransplant VOD is not a risk factor for TA-TMA.
VOD complicated 20 grafts. Two (10%) of the patients with VOD developed TA-TMA, whereas 23 (5.4%) developed TA-TMA out of 421 patients with no VOD (P = .83).
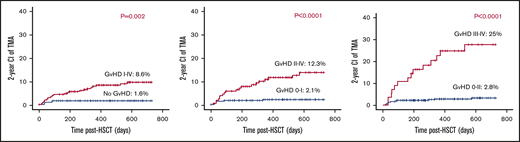
Noticeable rise in the risk for TA-TMA with higher grades of aGVHD.
Three patients failed to engraft after an MMUD cord transplant for IPEX syndrome, a haplo-HSCT for IL7Rα severe combined immune deficiency and an MUD BM for Fanconi anemia, so were considered nonevaluable for assessment of aGVHD.
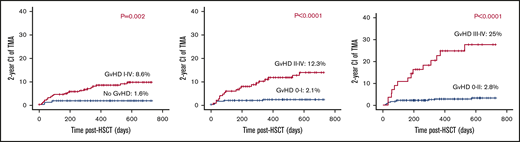
aGVHD grades I to IV, II to IV, and III to IV were recorded among 255 (58.2%), 155 (35.3%), and 56 (12.7%) of the 438 evaluable transplants. TA-TMA rates were significantly higher among cases with aGVHD grade II to IV (19/155) vs aGVHD grade 0 to I (6/283) (12.3% vs 2.1%; P < .001), and aGVHD grades III to IV (14/56) vs aGVHD grade 0 to II (11/382) (25% vs 2.8%; P < .0001; Figure 6).
Multivariate analysis on risk factors for TA-TMA
Variable P < .1 in the univariate analysis as risk factors for TA-TMA were included in the multivariate analysis. On multivariate analysis, the presence of active comorbidity, >1 transplant, and aGVHD grade III to IV were risk factors for TA-TMA (odds ratio [OR]: 5.1, 5.2, and 26.9; P = .002; P = .016; P < .0001, respectively), whereas the use of cyclosporine A (CSA)/tacrolimus-based GVHD prophylaxis and haploidentical transplant vs other HLA-matched grafts were not risk factors for TA-TMA (OR: 0.3, 1.1; P = .16 and P = .21; respectively) (Table 2).
Characteristics of patients who developed TA-TMA
Twenty-seven patients were considered to have TA-TMA and were treated accordingly. However, 2 of these patients did not meet Jodele et al 2016 criteria and thus were excluded from the current study. One of these patients was considered to have lung TA-TMA; however, postmortem biopsy showed organizing CMV pneumonia with no evidence of pulmonary microangiopathy. The second patient had grade III gut GVHD and disseminated treatment-refractory adenoviremia with BK virus hemorrhagic cystitis and developed seizures with radiological evidence of reversible leukoencephalopathy. During this time, she had only 3 out of 8 criteria for TA-TMA: proteinuria; hypertension; elevated lactate dehydrogenase (LDH). Subsequent renal biopsy confirmed BK virus nephritis as the cause of renal manifestations, with no evidence of microangiopathy or adenoviral infection.
Laboratory diagnostic criteria for TA-TMA, including raised LDH, proteinuria with associated hypertension, de novo thrombocytopenia, de novo anemia, and fragmented red cells, were reported among 22, 24, 24, 22, and 21 patients, respectively. Serum levels of C5b-9 were assessed among 15 patients: all had raised C5b-9 complex. Eight patients had tissue biopsy; TA-TMA was confirmed in 7 of them. Seven had confirmed renal (P6, P24) or lung microangiopathy (P1, P5, P7, P20, and P23). Supplemental Figure E1 shows a typical histopathology for lung TA-TMA. P24 developed proteinuria and hypertension with eye puffiness and lower-limb edema and had renal biopsy for probable autoimmune nephrotic syndrome; however, the biopsy showed microangiopathy. Twenty-one patients had genetic screening for genes involved in complement pathway with 3 having a confirmed mutation in the complement pathway: P3, P6, and P21. P21 had also a confirmed C5 polymorphism: c.2654G>A, known to be associated with resistance to eculizumab therapy. Supplemental Tables E3 and E4 and supplemental Figure E2 summarize the diagnostic criteria and characteristics of patients who developed TA-TMA. Of note, 16 out of 25 patients (62%) had either active aGVHD grade II to IV at time of TA-TMA (n = 8) or developed a flare-up of aGVHD on tapering or discontinuation of immune suppressive medications upon development of reversible leukoencephalopathy (P16, P19, P20) or after the development of TA-TMA: P8, P12, and P18. Moreover, P17 developed late-onset liver GVHD with reactivation of skin and gut, and P15 had a reactivation of upper gut GVHD prior to the onset of TA-TMA. The median time from onset or reactivation of aGVHD to development of TA-TMA among these 15 patients was 62 days (range: −11 days to 310 days). Twelve (12/16; 75%) patients had either steroid-refractory (n = 7) or steroid-dependent (n = 5) aGVHD.
Respiratory compromise requiring oxygen therapy (P12, P21, and P23) or assisted ventilation (P1, P2, P3, P4, P5, P9, P16, P17, P19, and P20) was recorded among 13 (52%) patients. Central nervous system manifestations, including encephalopathy or seizures, or both, were reported among 8 (32%) patients: P2, P4, P5, P14, P16, P20, P21, and P22. Renal compromise with raised creatinine with or without the need for replacement therapy was recorded among 6 (24%) patients: P12, P13, P16, P17, P20, and P23. Pleural or pericardial effusions, or both, were reported among 5 (20%) patients: P5, P6, P9, P10, and P12. Gastrointestinal bleeding occurred among 5 (20%) patients: P9, P16, P19, P21, and P22, whereas 3 (12%) patients developed cardiovascular instability requiring inotropes: P5, P19, and P20.
Moreover, 15 (60%) patients had severe disease requiring admission to pediatric intensive care unit. Complement blockers were used as first-line therapy in 18 patients: 16 received eculizumab and 2 received Coversin. Ten patients received defibrotide as either a first-line therapy (n = 6) or a second-line therapy (n = 4) after failure of complement blockers. Overall response rate to any treatment intervention was 52%: 13 out of the 25 patients with 3 of them (23%) having persistent hypertension or renal impairment as sequel postresolution of TA-TMA.
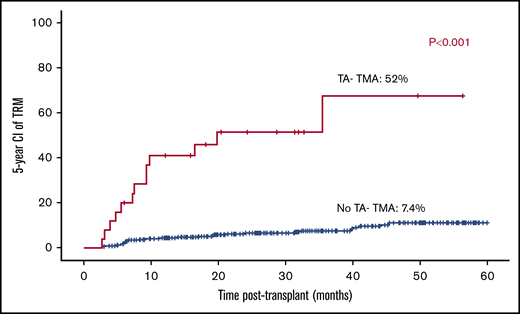
Overall TRM and effect of TA-TMA on TRM
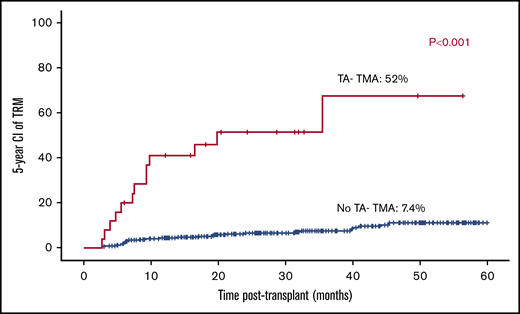
Overall, TRM was 9.7% (43/441). TRM was 5-fold higher among patients who developed TA-TMA (12/25; 48%) vs patients who did not develop TA-TMA (31/416; 7.4%; P < .001; Figure 7).
Among the TA-TMA group, 14 patients died at a median of 9.1 months (range: 2.7 to −35.5 months) posttransplant; 9 (64%) of them died due to pulmonary TA-TMA (n = 3) or sepsis complicating active aGVHD and TA-TMA (n = 3; P10, P17, P21), recurrence of HLH along with active aGVHD and TA-TMA (n = 1), disseminated adenoviremia with active TA-TMA (n = 1), respiratory syncytial virus–induced respiratory failure with active aGVHD, and uncontrolled TA-TMA (n = 1).
Other causes of death included disseminated atypical mycobacterial disease (n = 1), cerebral HLH (n = 1), toxoplasmosis (n = 1), and Aspergillus sepsis (n = 1). P19 died out of disease relapse and Aspergillus pneumonia. P19 had active TA-TMA at time of death. Eleven out of 16 (68%) patients with concomitant active aGVHD and TA-TMA died with TA-TMA being either the cause or one of the contributing factors for death vs 2 out of 8 (25%) who had no GVHD at time of the development of TA-TMA (P = .05) as shown in supplemental Table E4. P13 died of EBV-related lymphoproliferation. Steroid refractory, dependent, and responsive aGVHD was associated with a mortality rate of 66% (4/7 patients), 100% (4/4), and 50% (3/6), respectively.
Discussion
This is the first report to our knowledge that has identified comorbidity as a potential risk profile for a pediatric patient’s post-HSCT. Besides that, other risk factors, including aGVHD and a second transplant, were shown to be significant driving risk factors for TA-TMA. The identification of risk factors for the occurrence of TMA will conceivably allow early diagnosis and, perhaps, the timely introduction of therapeutic or preventive measurements.
Since the earliest report of TA-TMA in 1980,30 reports since 2004 showed a variable incidence of TA-TMA between 4% and 35%.16,31-36 Our report showed an incidence between 5% and 6%. Both centers in the United Kingdom reported similar incidences in the pediatric cohort. This consistency reflects the use of a uniform criterion to define TA-TMA and gives more insight into the true incidence of TA-TMA. Moreover, other studies that used similar criteria37 to define TA-TMA reported an incidence of between 7% and 8% among adults.16,35 To identify patients who developed TA-TMA, Uderzo et al21 used 6 laboratory criteria, including raised LDH >600 IU/L, red cell fragments >2%, unexplained drop of hemoglobin, and a drop in platelets <20 × 109/L after engraftment, elevated TA-TMA index (LDH/platelet) together with 3 clinical criteria: renal dysfunction with raised creatinine, central nervous system involvement, and consistent DIC, and reported an incidence of TA-TMA of 11% among adults and 13% among children. This report confirms the fact that use of a uniform criterion shows a more consistent incidence of TA-TMA.
Among the patient-derived risk factors, sex, and underlying disease did not influence the risk for TA-TMA, similar to data reported from previous studies.16,35,38 On the contrary, others reported increased risk of TA-TMA among females more than males, whereas underlying disease had no impact.21,39 In our cohort of patients, active comorbidity stood as a significant factor associated with TA-TMA. Although the pathophysiology of TA-TMA is not clearly understood, most studies reported endothelial injury to be the first and initiating step in the development of TA-TMA. Activation of endothelial cells to produce a procoagulant state leads to the activation of antigen-presenting immune cells and lymphocytes with production of multiple cytokines driving complement activation ending in microthrombus formation.40 Here, we defined active comorbidity using factors that are likely inducers of endothelial damage, including uncontrolled infections, the use of steroids to suppress active inflammation, cardiovascular instability, and pulmonary compromise.41-44
Reports from adults using hematopoietic cell transplantation-specific comorbidity index score to define comorbidity, however, failed to demonstrate a link between comorbidity and TA-TMA.16 However, this finding needs to be taken with caution in the pediatric population, where risk factors such as obesity, peptic ulcer, depression/anxiety, and coronary artery disease are seldom seen. Furthermore, the authors do not detail the risk profile for each patient. A current study in GOSH (N.B., R.E., P.A., G.L., R.C., P.V., and K.R., manuscript in preparation) demonstrated the failure of hematopoietic cell transplantation-specific comorbidity index score to be valid among 950 children who received a HSCT from 2006 to 2016.
Within transplant-related factors, factors that can potentially induce endothelial damage were analyzed. In our cohort, intensity of the conditioning regimen and use of TBI did not influence the development of TA-TMA with a risk between 5% and 6% with myeloablative, nonmyeloablative-conditioning regimen, or TBI-based conditioning. Nakamae et al45 showed in multivariate analysis that the use of Bu-based MAC conditioning was an independent risk factor for TA-TMA. Interestingly, in the same report, the risk of TA-TMA risk was 13% among all MAC and was not lowered (25%) among patients who received nonmyeloablative protocols: 77% being Flu-based conditioning. Eissner et al46 reported that Flu induces apoptosis, activation, alloreactivity of human endothelial cells and causes damage to dermal and alveolar epithelial cells. Flu is a major constituent of non-MAC, and the effect of Flu on endothelial cells was thought to contribute to the pathogenesis of TA-TMA after non-MAC.31,47 However, it was clear in the Elliott et al47 report that the patients who developed TA-TMA after Flu-based conditioning had undergone prior autologous transplantation and therefore were exposed to 2 transplant procedures. A subsequent conditioned HSCT was identified in our study cohort as a risk factor for TA-TMA with a risk factor of 15% vs 5% for patients who received only 1 graft. This is similar to previous studies in adults with a risk ranging between 12%16,35 and 52%30 with subsequent transplant vs an average of 7% in patients receiving only 1 graft.16 The effect of TBI has been controversial with some reports showing increased incidence of TA-TMA,21,39 whereas others including ours showing no effect on the development of TA-TMA.16,35
The use of a mismatched graft was considered a potential risk factor for TA-TMA in adults.21,45,48,49 We reported a significantly higher incidence of TA-TMA of 17% with haploidentical HSCT vs <10% among other donor sources. Within this group, grade III to IV aGVHD and the presence of active comorbidity were the main driving risk factors for increased TA-TMA in our study.
The occurrence of aGVHD was the most important risk factor to TA-TMA in our series. These results are concordant with other studies that reported a strong link between GVHD and TA-TMA. Some considered the link as being related to the use of more immunosuppressive medications, in particular, CSA or high levels of tacrolimus, combination of CSA with sirolimus as prophylaxis, or therapy for GVHD, 18,45,50, with existing guidelines recommending discontinuation of calcineurin inhibitors as the main intervention after initial diagnosis.1,7,40 The mechanism appears to be partly due to decreased levels of nitric oxide and vascular endothelial growth factor.40 In vitro studies have shown that both CSA and tacrolimus + sirolimus increased expression of intracellular adhesion molecule-1 with CSA alone having a further detrimental effect on the endothelium through increasing reactivity of extracellular matrix to platelets inducing a procoagulant status.51
However, 2 factors seem not to support this hypothesis. First, in previous reports, the withdrawal of calcineurin inhibitors failed to reverse or even to halt the process of TA-TMA.16,21,31 Uderzo et al21 demonstrated clearly that the withdrawal of immunosuppressive medications did not prevent mortality, where 11 out of 14 (78%) patients who stopped immunosuppressive therapy died vs 21 out of 49 (42%) patients who continued on immunosuppression. Within another report on 22 patients with TA-TMA, 18 of them had either discontinued CSA or reduced the dose in addition to other therapeutic measures: plasma infusion and/or therapeutic plasma exchange. A response was only recorded among 5 out of 18 patients. Of note, nonrelapse mortality was 77% (14 out of 18 patients) with GVHD being the cause of death among 9 of them. A recent report by Li et al16 showed similar results where 60 patients with TA-TMA either discontinued CSA or tacrolimus or sirolimus (31 patients) or shifted from one to another (29 patients) with no evidence that withdrawal of either calcineurin inhibitors or sirolimus was beneficial, with a clear demonstration that GVHD grades III to IV were the main driving factor for TA-TMA. Second, in a pilot study carried out by Ringdén et al including 24 HSCT patients, comparing the use of tacrolimus/sirolimus to use of CSA with or without methotrexate, none of the patients developed grade III to IV aGVHD or TA-TMA. Hence, being on calcineurin inhibitors or sirolimus with the absence of high-grade GVHD, TA-TMA was not reported.52 In our series, when using multivariate analysis, we noted that neither the use of calcineurin inhibitors nor the use of mismatched grafts, including haplo-HSCT, drove the development of TA-TMA, with aGVHD grades II to IV and III to IV being the main risk factors for TA-TMA. A growing body of literature supports the role of endothelial dysfunction in the development of GVHD53-56 with evidence of complement activation as a downstream effect of endothelial dysfunction.57,58 Recent studies in adults showed a strong association between aGVHD grades III to IV and renal TA-TMA,54 steroid refractory GVHD and TA-TMA,58 and gastrointestinal aGVHD and TA-TMA38 with dismal outcomes across most of the reports. In our series, more than half of the patients who were diagnosed with TA-TMA with concomitant aGVHD died. This is also similar to previous reports from adults where Zhang et al38 reported poor survival of 52% (n = 50) among patients who developed TA-TMA vs 77% in patients who had only aGVHD (n = 83) and 86.6% among patients who neither developed aGVHD nor TA-TMA and aGVHD (n = 67) with higher nonrelapse mortality but low risk of relapse. Another group reported a drop in 90-day survival among patients with TA-TMA from 96% in patients with no documented driving factor to 50% in patients with concomitant GVHD. All these studies, including ours, demonstrate that vascular GVHD might be a specific form of TA-TMA that requires intensified treatment of GVHD to control TA-TMA rather than discontinuation of immune suppressive medications, which might worsen GVHD leading to increased mortality.
To date, current therapeutic recommendations for TA-TMA are controversial with no approved medicine by regulatory authorities. Therapeutic plasma exchange, in addition to being invasive, has shown no benefit in treating adults with TA-TMA2,19,31 possibly due to the lack of autoantibodies against factor H or other endothelial proteins.40 With more insight into TA-TMA and evidence of terminal complement activation, complement blockers like eculizumab represent an appealing therapeutic intervention for TA-TMA; however, a failure rate of 48% was recorded in our series, which is similar to other reports with 30% to 40% response failure.59,60
In our studied cohort, the choice on the use of complement blockade vs defibrotide was made based on the accessibility to treatment, the analysis of complement activation, the presence of bleeding, and the center experience.
In patients treated at GOSH, P10 to P25 had evidence of complement activation as shown by elevated sC5b-C9. With access to a complement blockade, either eculizumab or Coversin was used among all patients except P24. P24 was the only patient from GOSH whose complement was not checked at the start of proteinuria. Diagnosis of TA-TMA came after renal biopsy confirming microangiopathy. On the contrary, the GNCH BMT center with easier access to defibrotide and previous experience with the use of defibrotide therapy in TA-TMA29 used defibrotide in most patients who developed TA-TMA.
When there is evidence of complement activation, the use of complement blockade should be considered an option to stop the evolution of TA-TMA. However, complement activation is not the sole player for the development and evolution of TA-TMA. Defibrotide, which has broad-spectrum anti-thrombotic, anti-ischemic, and anti-inflammatory activity, may protect the endothelium from damage associated with TA-TMA. Conversely, if TA-TMA is associated with bleeding, then defibrotide may be contraindicated.
A study by IEWP and PDWP EBMT on the use of defibrotide in TA-TMA recorded a resolution rate of 77% among both children (17/22) and adult (13/17) cohorts.29
A recent multicenter retrospective study on behalf of IEWP and PDWP EBMT (P.A., G.L., R.C., P.V., and K.R., submitted for publication 2020) including 29 centers, 80 allo-HSCTs, and 6 auto-HSCTs, 55 patients (68%) received eculizumab as a first-line measure (elevated sC5b-9 were checked in only 41 patients). At 6 months from the start of therapy, the cumulative incidence of TA-TMA resolution was 41%, and overall survival was 50%.
In order to assess the true benefit of defibrotide in TA-TMA patients, a prospective multicenter study is required to allow accurate comparison between defibrotide and eculizumab/Coversin to determine which patients are most likely to benefit from treatment and guide treatment dose and duration.
Given the severity of TA-TMA and the noted high rate of mortality recorded in all studies to date, this requires consideration of potential prophylactic measures to prevent the occurrence of TA-TMA. One of the most appealing prophylactic medications is defibrotide. Defibrotide is a mixture of single-stranded oligonucleotides with endothelial protection properties mediated via inhibition of tumor necrosis factor-α and its pro-fibrinolytic, antithrombotic, anti-inflammatory, and thrombolytic properties. It is US Food and Drug Administration approved for the treatment of VOD, another endothelial damage complication of HSCT.40 The use of defibrotide the day prior to conditioning has been demonstrated to effectively provide protection against the development of VOD in high-risk patients.61 In vitro, defibrotide protects against endothelial injury induced by Flu and calcineurin inhibitors.46,51 A trial of defibrotide prophylaxis in patients at high risk for development of TA-TMA is currently underway in the United States (NCT#03384693).
To sum, our study has demonstrated a profile for pediatric patients undergoing HSCT who are at greater risk of developing TA-TMA. We have shown that higher-grade aGVHD, multiple transplants, and comorbidity as being the main drivers for pediatric TA-TMA. Given the nonavailability of effective treatments and high nonrelapse mortality associated with TA-TMA, identification of patients at risk and considering prophylactic measures may be worthwhile. The link between GVHD and TA-TMA demonstrated in our study and other recent reports identifies a particular form of TA-TMA that might represent a vascular GVHD. One alternative approach might be to substitute calcinuerine inhibitors with other modalities of immunosuppression; however, with this approach, it is paramount that good control of GVHD is achieved or TA-TMA may be exacerbated.
Acknowledgments
The authors acknowledge support for this work by the MRC/ESPRC Newcastle Molecular Pathology Node and NIHR BRC GOSH.
Authorship
Contribution: R.E. designed the research and wrote the manuscript; R.E. and J.S. analyzed the data; G.L., S.-H.L., A.B., Z.N., G.O., N.B., R.C., K.R., P.A., S.O., T.F., A.J.C., R.S., and A.W. were actively involved in patients' care, collected data, and helped in designing the research; W.Q., C.B., M.S., A.R.G., and P.V. checked data and worked on editing the writing of the manuscript; H.Y. and B.D., from pharmacy, wrote and analyzed data on defibrotide and eculizumab; D.K., S.G., and P.W., from the genetics laboratory, ran genetics on complement; and S.A. revised the histopathology slides.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Reem A. Elfeky, Great Ormond Street (GOS) Hospital for Children NHS Foundation Trust, University College London GOS Institute of Child Health, and NIHR GOSH BRC, 30 Guilford St, London WC1N 3JH, United Kingdom; e-mail: reem.elfeky@gosh.nhs.uk and r.elfeky@ucl.ac.uk.