

Key Points
No occlusion was found without α-granules in both in vitro and in vivo models (Nbeal2−/− mice), but occlusion occurred after rescue.
Platelet α-granules are necessary for rapid platelet accumulation.

Abstract
von Willebrand factor (VWF) is essential for the induction of arterial thrombosis. In this study, we investigated the critical role of platelet VWF in occlusive thrombosis formation at high shear in mice that do not express platelet VWF (Nbeal2−/−). Using in silico modeling, in vitro high-shear microfluidics, and an in vivo Folts model of arterial thrombosis we reproduced the platelet dynamics that occur under pathological flow in a stenosed vessel. Computational fluid dynamics (CFDs) simulated local hemodynamics in a stenosis based on arterial geometries. The model predicted shear rates, time course of platelet adhesion, and time to occlusion. These predictions were validated in vitro and in vivo. Occlusive thrombosis developed in wild-type control mice that had normal levels of plasma VWF and platelet VWF in vitro and in vivo. Occlusive thrombosis did not form in the Nbeal2−/− mice that had normal plasma VWF and an absence of platelet VWF. Occlusive thrombosis was corrected in Nbeal2−/− microfluidic assays by the addition of exogenous normal platelets with VWF. Combining model and experimental data, we demonstrated the necessary requirement of platelet VWF in α-granules in forming an occlusive thrombus under high shear. These results could inspire new pharmacological targets specific to pathological conditions and prevent arterial thrombosis.
Introduction
Arterial thrombosis is one of the most frequent causes of morbidity and mortality in Western society.1 The rapid capture and accumulation of circulating platelets at atherosclerotic rupture sites can cause thrombotic arterial occlusion preceding acute myocardial infarction or stroke.2,3 Under normal physiological conditions, typical shear rates of 15 to 1600 s−1 occur in the arterial vasculature.4 However, atherosclerotic stenosis can increase local shear rates dramatically to 5000 to 400 000 s−1,5 generating high hemodynamic forces and a prothrombogenic environment. Currently, there are no clinical therapeutics that can unambiguously target platelets at these pathological forces at the site of occlusive thrombus formation. Occlusive thrombus formation under these high-shear conditions can be described as a 3-phase process of (1) initial platelet attachment to a thrombogenic surface (lag phase); (2) accelerated growth (rapid platelet accumulation [RPA] phase); and (3) thrombus stabilization or cessation of blood flow (occlusion phase).6
Platelet granules and their contents have a central role in controlling the balance between hemostasis and thrombosis,7-9 by driving the 3 phases of thrombotic occlusion. Individuals who lack or have abnormally low platelet α-granules (gray platelet syndrome [GPS]); dense granules (Hermansky-Pudlak syndrome [HPS]); or both (αδ-storage pool disease) display variable pathologies and bleeding diathesis.8,10,11 Mouse strains that lack α-granules (Nbeal2−/−)12,13 or dense granules (ruby eye)14,15 or have defective α- and dense-granule exocytosis (VAMP−/−),15 have also been identified and echo similar pathologies and varying bleeding tendencies, as seen in humans. Although studies involving these mouse strains and their granule cargo vary among the different thrombosis models, it has been proposed that dense granules, which store and secrete ADP, ATP, serotonin, and calcium, promote primary aggregation by activating and recruiting platelets to injury sites.7,8 A deficiency of dense granules or secretion of their contents typically results in a diminished secondary aggregation response of surrounding platelets and delayed or attenuated initial thrombus formation in vivo.16 Dense granules initiate and support platelet aggregation, and α-granules store and secrete adhesive proteins, such as von Willebrand factor (VWF) and fibrinogen, that mediate thrombus growth and stability via platelet-platelet interactions.17 Like dense granule defects, an absence of α-granules (Nbeal2) also produces various pathologies and bleeding.12,16,18-23 In tail-bleeding assays, Nbeal2−/− mice exhibit increased blood loss, suggesting a deficiency in hemostasis. However, in stroke models, an absence of Nbeal2 causes no intracranial hemorrhage.13,19 In ferric chloride and laser injury models, Nbeal2−/− mice have impaired thrombus growth and stability, although the rate of platelet activation is normal.12,13,24 The observations in Nbeal2−/− mice are α-granule specific, as that dense granules and VWF in the plasma and endothelium are normal.
Previous studies using Nbeal2−/− mice demonstrate impaired thrombus formation under physiological conditions.12,13,24 Although these critical studies have demonstrated a role for α-granules in hemostasis and thrombosis, occlusive thrombus formation under pathological high-shear conditions remain unknown. α-Granules contain VWF and coagulation factors, including fibrinogen, PF4, thrombospondin, and factor V. VWF is the dominant protein that mediates platelet aggregation and thrombosis under high shear.25 Coagulation factors play a lesser role under high shear, because they are likely to be transported away from the stenosis before the coagulation cascade is completed. Although fibrinogen plays a more active role at low shear (<1000 s−1), there are studies showing that fibronectin can promote platelet aggregation and thrombus growth.26 However, fibronectin concentration in α-granules (0.19 mg/mL) is smaller than plasma concentration (0.30 mg/mL),27 which in turn have less effect on thrombosis in the absence of α-granules.
VWF is synthesized and stored in α-granules of platelets and in the Weibel-Palade bodies of endothelial cells. High-molecular-weight multimers and ultralarge multimers (ULMs) of VWF are constitutively secreted into the blood plasma,28,29 and are cut to smaller multimer forms of VWF by the metalloprotease ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type-1 motif, member 13). The concentration of VWF in normal human plasma is ∼10 μg/mL, whereas VWF in platelet α granules are present at the much higher concentration of 500 μg/mL.27 Under physiological conditions, VWF does not interact with the endothelium or other platelets; however, when exposed to high shear rates, tethered VWF elongates and exposes its A1 and A2 binding domains.30,31 The exposure of the A1 domain enables rapid A1-platelet interactions that support rapid platelet-platelet and platelet-endothelial interactions32 at high-shear rates,33 independent of activation.31 In response to high shear stress, high-molecular-weight and ultra-large VWF multimers have increased platelet binding capacity and are more thrombogenic.34-36
Although the involvement of VWF in thrombus formation is well established,37-43 the source of VWF at different stages of formation is less clear. Plasma and endothelial VWF concentrations are sufficient for initial platelet adhesion during the lag phase.39,41,44 Theoretical calculations of bond strength and growth rate indicate that the concentration of local VWF must be much greater than plasma concentrations to capture the passing platelets at the observed rate.35 Further, the lag time of slow aggregation could be explained by the equivalent time of shear activation of mural platelets to release their α-granule VWF before RPA can occur.13 Based on this evidence, we hypothesized that platelet α-granule release of VWF and other contents is necessary for RPA to proceed to occlusion beyond the initial platelet attachment to the subendothelium.31,35,41
In the present study, using in silico fluid dynamic simulations, in vitro microfluidics, and in vivo models of arterial thrombosis, we investigated thrombus formation, with and without platelet α-granule release, at high shear rates. To identify the critical role of platelet VWF in high shear occlusive thrombosis we used Nbeal2−/− mice that are deficient in α-granules and therefore do not release platelet VWF and other contents, but have normal levels of plasma VWF similar to WT.12
Understanding the time scales, shear rates, and conditions that lead to occlusive thrombosis under pathological conditions could lead to better targeted pharmacological inhibitors for the prevention of arterial thrombosis without bleeding side effects.
Materials and methods
Computational fluid dynamics analysis
Computational fluid dynamics (CFD) analysis was used to predict shear-rate distribution within microfluidic and carotid artery geometries. Simulations were performed using Ansys 19.1 (Ansys Inc, Canonsburg, PA). Whole blood flowing at normal and high shear rates was assumed to be Newtonian fluid of 3.5 cP, and flow was presumed to be laminar, incompressible, steady continuum, and isothermal because of the Reynolds number of the experiments. The microfluidic chamber was modeled as a rectangular channel with a total pressure inlet of 4.8 mm Hg and a uniform 0-pressure (gauge) outlet. The no-slip boundary condition was applied to the walls (Figure 1A). A stenosed mouse carotid artery was modeled as a 60% diameter constriction of a cylindrical vessel, and a 0.25 mL/min flow rate was applied at the inlet, with 0 pressure at the outlet (Figure 1B). Mesh convergence was achieved at 3.5 million (microfluidics) and 4 million (mouse carotid artery) tetrahedral cells, yielding a residual error of 10−9 for each model, respectively.
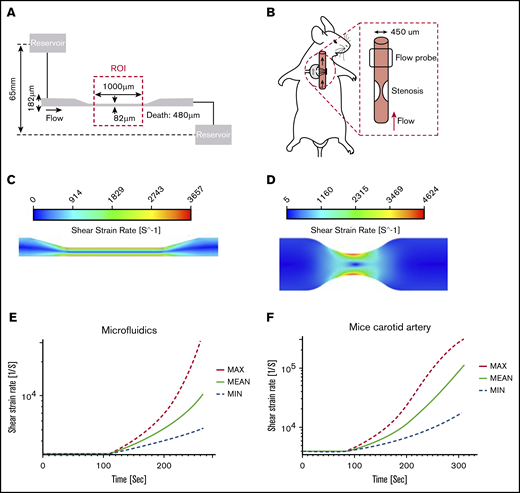
Hydrodynamic flow characteristics through a high-shear microfluidic device and stenosed mouse carotid artery. (A) Geometry of the microfluidic model with an experimental stenotic section. (B) In vivo experiments showing a stenotic region relative to flow direction and flow probe placement. (C) CFD simulations of the microfluidic device. Blood flowed from left to right. The color contour shows shear strain rate distribution. The solution showed a maximum shear strain rate of >3500 s−1 in the stenosis region, whereas upstream and downstream values were <1000 s−1. (D) CFD simulation in a stenosed mouse carotid artery. The large velocity gradient at the stenotic apex led to initial shear rates that exceeded 4000 s−1. (E-F) Thrombus growth was predicted by using a high-shear thrombosis model developed by Mehrabadi et al.45 Shear strain rate estimation is shown over time during high-shear thrombosis formation in the microfluidic assay and mouse carotid artery. Min and Max denote the lower and upper confidence limits of the model, respectively.
Hydrodynamic flow characteristics through a high-shear microfluidic device and stenosed mouse carotid artery. (A) Geometry of the microfluidic model with an experimental stenotic section. (B) In vivo experiments showing a stenotic region relative to flow direction and flow probe placement. (C) CFD simulations of the microfluidic device. Blood flowed from left to right. The color contour shows shear strain rate distribution. The solution showed a maximum shear strain rate of >3500 s−1 in the stenosis region, whereas upstream and downstream values were <1000 s−1. (D) CFD simulation in a stenosed mouse carotid artery. The large velocity gradient at the stenotic apex led to initial shear rates that exceeded 4000 s−1. (E-F) Thrombus growth was predicted by using a high-shear thrombosis model developed by Mehrabadi et al.45 Shear strain rate estimation is shown over time during high-shear thrombosis formation in the microfluidic assay and mouse carotid artery. Min and Max denote the lower and upper confidence limits of the model, respectively.
Arterial thrombosis model for simulating occlusion time
Time to occlusion in a stenotic microfluidic test section or mouse carotid artery was predicted using a quantitative, empirical high-shear thrombosis model.45
Channel height/diameter was updated every second as thrombus growth increased in both models.
Mice
Nbeal2−/− and WT control mice were generated from Nbeal2+/− mice on a C57/bl6 background with a targeted deletion region between exons 4 and 11 of Nbeal2 (from strain B6;129S5-Nbeal2tm1Lex/Mmucd).12 An equal number of 12- to 20-week-old male and female mice were used. All animal experiments were approved by the university animal care and use committees.
High-shear microfluidic model of stenosis
Microchannels with a stenotic region were used for in vitro studies of whole-blood occlusion at pathological shear39 (Figure 1A). Whole blood was collected from the inferior vena cava of Nbeal2−/− and WT mice into heparin anticoagulant (15 USP units/mL), and platelets were fluorescently labeled with DiOC6 (3,3′-dihexyloxacarbocyanine iodide; Invitrogen). Whole blood was perfused through the microfluidic device at a constant upstream pressure head of 65 mm on immobilized type 1 fibrillar collagen (Chronolog, Havertown, PA). Platelet adhesion and occlusion were captured in real time with an ORCA Flash4 CMOS camera (Orca‐ER; Hamamatsu, Hamamatsu City, Japan) and CellSens software (Olympus, Center Valley, PA), with images being acquired every 1 second for the duration of the experiment. Occlusion time was defined for each series when centerline red blood cell (RBC) velocities decreased to less than 100 μm/s.
Folts-like arterial stenosis model
In vivo thrombus formation under pathological shear was assessed using the Folts-like model of arterial thrombosis.46 A 6.0 silk suture was tied around the common carotid artery until blood flow rate was reduced to ∼50%, as monitored with a flow probe (Transonic Systems, Ithaca, NY), to create a stenosis region and a high-shear-stress environment. The carotid vessel under the stenosis was gently crushed with forceps, to expose the subendothelial surface, and blood flow was continuously monitored. Blood flow data were acquired with Powerlab and analyzed using LabChart Pro version 8.1 (ADInstruments, Colorado Springs, CO). Occlusive thrombus formation occurred when blood flow decreased to 0. After the experiments, the carotid artery including the injury site was dissected and fixed in neutral-buffered formalin fixative overnight. Tissue was embedded in paraffin, sectioned, and stained either with hematoxylin and eosin or by the Carstairs method (Electron Microscopy Sciences, Hatfield, PA). Histology images were captured with an Olympus BX42 microscope and Olympus DP72 digital camera with CellSens software (Olympus). Thrombus structure and composition were assessed where platelets stained blue/purple, fibrin stained red, red blood cells stained yellow, and collagen appeared blue.
Analysis
Image analysis was performed with FIJI/ ImageJ 1.51n software (National Institutes of Health, Bethesda, MD), and the data were processed with Matlab (Mathworks, Natick, MA). Data analysis was performed with Prism 7 (GraphPad Software, San Diego, CA).
Statistical significance (P < .05) was determined by the unpaired Student t test or by a 1-way analysis of variance with the Dunnett post hoc test for multiple comparisons.47
Results
Hemodynamic flow characteristics through a high-shear microfluidic device and stenosed mouse carotid artery
Flow characteristics through a simulated high-shear microfluidic device (Figure 1A) and stenosis in a mouse carotid artery (Figure 1B) were quantified through CFDs, and a high-shear thrombosis growth model was used to predict the occlusion time.45 Our model was simplified by assuming constant flow rate and upstream pressure as an end point to predict occlusion time.
In microfluidic simulations, the maximum initial shear rate occurring at the stenotic region of the channel was >3500 s−1, and the nonstenotic upstream and downstream regions were <1000 s−1, corresponding to normal physiological arterial wall shear rates (Figure 1C).
A stenosis with axisymmetric geometry was created to compute shear rate in a mouse carotid artery model. Simulations were performed with a 60% stenosis to create an ∼50% reduction in flow rate based on previous studies.5 A maximum initial shear rate of >4500 s−1 at the apex of the stenosis was predicted. No flow separation was seen distal to the stenosis in this mouse artery because of the low Reynolds number associated with the small vessel of a mouse (Figure 1D).
A comparison between the microfluidics model and the mouse carotid artery stenosis model showed that the shear rate distribution in both stenosis models was very similar (Figure 1C-D). To determine occlusion times in our simulations, a constant flow rate was assumed. Occlusion was defined as when the inlet pressure increased to 4.8 mm Hg with the microfluidic device, and when the arterial pressure proximal to the stenosis reached 100 mm Hg in the mouse modified-Folts model. Microfluidic device simulations predicted an occlusion time of 264 ± 65 seconds. The mouse carotid artery with 60% stenosis was predicted to have an occlusion time of 305 ± 77 seconds (∼5 minutes). Based on predicted occlusion times, we made sufficient observation windows for microfluidics of 8 minutes and for the mouse carotid artery of 12 minutes. The shear rate and simulated thrombus growth built up in the microfluidic channel and stenosed mice artery, as shown in Figure 1E-F, where the shear rate was calculated to increase as high as 10 000 and 100 000 s−1, respectively.
Platelet VWF released by shear is necessary for forming subsequent occlusive thrombi
To isolate the contributions of platelet VWF to the formation of occlusive thrombus at high shear, we performed perfusion assays on type 1 collagen in microfluidic channels that mimic the stenosis of arteries using WT and Nbeal2−/− mouse whole blood. The hemodynamics previously quantified through CFDs were recapitulated in our device. Whole blood was perfused through the device under a constant upstream pressure of 4.8 mm Hg to create wall shear rates of ∼3500 s−1 at the stenosis and <1000 s−1 at the nonstenosis regions (Figure 1C). RBC centerline velocities distal to the thrombi <100 μm/s were used to define the occlusion time.
WT mice formed characteristically normal thrombi at the stenotic test section (Figure 2A). An initial lag phase of continuous adhesion and aggregation (see adherent platelets at 1 minute) was proceeded by an RPA phase characterized by an increase in thrombus size and intensity (Figure 2C). WT mice occluded the microfluidic stenosis test region within 326 ± 25 seconds (Figure 2C) as measured by RBC flow (n = 6; P < .001). In contrast, Nbeal2−/− mouse whole blood exhibited a normal lag phase adhesion of platelets to collagen within the first minute, but then no RPA phase or occlusion was observed during the next 8 minutes of follow-up (Figure 2B-C; n = 6; P < .001).
![Plasma VWF is sufficient for initial adhesion of platelets to collagen, whereas platelet VWF released locally is necessary for forming subsequent occlusive thrombi. Representations of WT and Nbeal2−/− platelet adhesion and subsequent occlusion during the 8-minute perfusion period. The progressive accumulation of platelets is observed over time. (A) WT mice blood showed rapid and significant platelet adhesion and aggregation leading to occlusion. (B) Nbeal2−/− mice blood did not form occlusive thrombi during the 8-minute observation period. Occlusive thrombi formed after the addition of WT whole blood (Rescue) to Nbeal2−/− platelets adherent to collagen. The 1-mm stenosis region is marked with a red line under the image. The ROI appears blank before the adhesion of platelets (green; 3,3′-dihexyloxacarbocyanine iodide [DiOC6]). Flow direction is from left to right. The bar represents 1000 μm. (C) Normalized fluorescent intensities comparing thrombus growth and occlusion in control WT (n = 6), Nbeal2−/− (n = 6), and Rescue (n = 3) channels. Initial lag phase quickly proceeded to RPA, demonstrated by a rapid increase in thrombus size and intensity at ∼100 seconds for WT whole blood and at 515 ± 47 seconds for Nbeal2−/−+WT whole blood. Occlusion occurred at the end of the plot for WT and Nbeal2−/−+WT whole blood. No occlusion was observed with Nbeal2−/− whole blood alone. The arrow denotes the time point when WT whole blood was added to Nbeal2−/− channels. All images were postprocessed and averaged to compare the intensity levels in the test sections. (D) Occlusion times in the microfluidic device for WT (326 ± 25 seconds), Nbeal2−/− (no occlusion), and Nbeal2−/−+WT (166 ± 48 seconds). All 3 occlusion times were significantly different from each other. ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/14/10.1182_bloodadvances.2020002117/4/m_advancesadv2020002117f2.png?Expires=1767899350&Signature=M0XCSxZ7MMuA7GYSJMHkB2iipe8uO8jS6F-Ramq9w2zhPuo7HMnxENmD~fyI7agXFwf40hjFbrH6v8q4jOpR4E3Bha-kf-8O9FCBkikGrRMmHRTz2C6IeQOsBWC-xOkKhWd0pXl6u3wA4G5vh~ya6~Rcoh0B8OT7JK6gMOftz3pIdeSNoF4LTqQJhaoZBTHqXR4T0WzCPO2Ryi9-CddoEg0S2hskMfMoB2-IUSgg15dy6u7hHDDa8NN-ms8dMDOEQznwbd-uPbyIRAFqJnSxY6pXj8lQ8G76qq6zgSXUHl-47Cxb1tTW05QWf-1PgwA8dfahKiCe-mqiwoCV4DI5cw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Plasma VWF is sufficient for initial adhesion of platelets to collagen, whereas platelet VWF released locally is necessary for forming subsequent occlusive thrombi. Representations of WT and Nbeal2−/− platelet adhesion and subsequent occlusion during the 8-minute perfusion period. The progressive accumulation of platelets is observed over time. (A) WT mice blood showed rapid and significant platelet adhesion and aggregation leading to occlusion. (B) Nbeal2−/− mice blood did not form occlusive thrombi during the 8-minute observation period. Occlusive thrombi formed after the addition of WT whole blood (Rescue) to Nbeal2−/− platelets adherent to collagen. The 1-mm stenosis region is marked with a red line under the image. The ROI appears blank before the adhesion of platelets (green; 3,3′-dihexyloxacarbocyanine iodide [DiOC6]). Flow direction is from left to right. The bar represents 1000 μm. (C) Normalized fluorescent intensities comparing thrombus growth and occlusion in control WT (n = 6), Nbeal2−/− (n = 6), and Rescue (n = 3) channels. Initial lag phase quickly proceeded to RPA, demonstrated by a rapid increase in thrombus size and intensity at ∼100 seconds for WT whole blood and at 515 ± 47 seconds for Nbeal2−/−+WT whole blood. Occlusion occurred at the end of the plot for WT and Nbeal2−/−+WT whole blood. No occlusion was observed with Nbeal2−/− whole blood alone. The arrow denotes the time point when WT whole blood was added to Nbeal2−/− channels. All images were postprocessed and averaged to compare the intensity levels in the test sections. (D) Occlusion times in the microfluidic device for WT (326 ± 25 seconds), Nbeal2−/− (no occlusion), and Nbeal2−/−+WT (166 ± 48 seconds). All 3 occlusion times were significantly different from each other. ***P < .001.
Plasma VWF is sufficient for initial adhesion of platelets to collagen, whereas platelet VWF released locally is necessary for forming subsequent occlusive thrombi. Representations of WT and Nbeal2−/− platelet adhesion and subsequent occlusion during the 8-minute perfusion period. The progressive accumulation of platelets is observed over time. (A) WT mice blood showed rapid and significant platelet adhesion and aggregation leading to occlusion. (B) Nbeal2−/− mice blood did not form occlusive thrombi during the 8-minute observation period. Occlusive thrombi formed after the addition of WT whole blood (Rescue) to Nbeal2−/− platelets adherent to collagen. The 1-mm stenosis region is marked with a red line under the image. The ROI appears blank before the adhesion of platelets (green; 3,3′-dihexyloxacarbocyanine iodide [DiOC6]). Flow direction is from left to right. The bar represents 1000 μm. (C) Normalized fluorescent intensities comparing thrombus growth and occlusion in control WT (n = 6), Nbeal2−/− (n = 6), and Rescue (n = 3) channels. Initial lag phase quickly proceeded to RPA, demonstrated by a rapid increase in thrombus size and intensity at ∼100 seconds for WT whole blood and at 515 ± 47 seconds for Nbeal2−/−+WT whole blood. Occlusion occurred at the end of the plot for WT and Nbeal2−/−+WT whole blood. No occlusion was observed with Nbeal2−/− whole blood alone. The arrow denotes the time point when WT whole blood was added to Nbeal2−/− channels. All images were postprocessed and averaged to compare the intensity levels in the test sections. (D) Occlusion times in the microfluidic device for WT (326 ± 25 seconds), Nbeal2−/− (no occlusion), and Nbeal2−/−+WT (166 ± 48 seconds). All 3 occlusion times were significantly different from each other. ***P < .001.
To further demonstrate that normal plasma VWF alone is not enough to form occlusive thrombus, we then reperfused WT whole blood into the nonoccluded Nbeal2−/− channels (Figure 2B). Perfusion of WT led to immediate RPA of the earlier collagen-bound Nbeal2−/− platelets, and occlusion was observed 166 ± 48 seconds later (Figure 2D). Collectively these results conclusively demonstrate that plasma VWF supports the primary adhesion of platelets to collagen during a lag phase, whereas platelet VWF is essential for RPA and occlusive thrombus formation at high shear rates.
Platelet VWF release induces localized formation of an occlusive thrombi
Localized release of VWF from activated platelets could induce a localized formation of occlusive thrombi. Using identical microfluid channels and gravity-driven flow, Griffin et al48 found that localized thrombi formed with human blood. To investigate whether the same effect would occur with mouse blood, we split the region of interest (ROI) into 3 ROIs (Figure 3A) and compared the intensity level of each ROI. The final intensity level in ROI 1 (upstream) was significantly higher than that in the other 2 ROIs for WT and Rescue (Figure 3B; P < .01), but there was no significant difference between ROIs for Nbeal2−/−. The intensity level of each ROI during the perfusion time (Figure 3C-D) showed a sudden increase in intensity in ROI 1 for WT (80 seconds) and Rescue (20 seconds), but only a small increase in intensity in ROIs 2 and 3. Thus, RPA occurs in the upstream part of the stenosis (333 μm) and serves as the dominant location for the channel occlusion in the microfluidic system.

Platelet VWF induces localized formation of occlusive thrombi. (A) Three ROIs were delimited for the microfluidic channel test section. (B) Normalized fluorescence intensity level of each ROI after an 8-minute perfusion for WT (n = 6) and Nbeal2−/− (n = 3) and 5 minutes for Rescue (n = 3). ROI 1 showed a significantly higher intensity level with WT and Rescue, compared with ROIs 2 and 3. **P < .01; ***P < .001. There was no significant increase in ROI 1 with Nbeal2−/− blood. (C-D) Intensity levels of each ROI during the perfusion time. The arrow denotes the time point when WT whole blood was added to Nbeal2−/− channels. The intensity level of ROI 1 rapidly increased after 80 seconds in the WT and 20 seconds in the Rescue mice. ns, not significant.
Platelet VWF induces localized formation of occlusive thrombi. (A) Three ROIs were delimited for the microfluidic channel test section. (B) Normalized fluorescence intensity level of each ROI after an 8-minute perfusion for WT (n = 6) and Nbeal2−/− (n = 3) and 5 minutes for Rescue (n = 3). ROI 1 showed a significantly higher intensity level with WT and Rescue, compared with ROIs 2 and 3. **P < .01; ***P < .001. There was no significant increase in ROI 1 with Nbeal2−/− blood. (C-D) Intensity levels of each ROI during the perfusion time. The arrow denotes the time point when WT whole blood was added to Nbeal2−/− channels. The intensity level of ROI 1 rapidly increased after 80 seconds in the WT and 20 seconds in the Rescue mice. ns, not significant.
An absence of platelet VWF prevents occlusive thrombus formation in the Folts-like arterial stenosis model
To investigate the contribution of platelet-derived VWF in promoting occlusive thrombus formation in vivo, we used WT and Nbeal2−/− mice in a modified Folts arterial model.15 A stenosis was made by decreasing blood flow in the carotid artery by ∼50% and generating a high-shear-stress zone. Occlusion occurred in all WT mice at an average time of 330 seconds after deendothelization of the stenosed vessel. In contrast, no occlusive thrombus formation was observed in the carotid arteries of all Nbeal2−/− mice (Figure 4A-B; n = 7; P < .0005).
![An absence of platelet VWF prevents occlusive thrombus formation in the modified Folts arterial stenosis model. (A) Mean blood flow through the carotid artery was reduced to 50% by making stenosis and subsequently crush injury was induced. WT mice consistently formed an occlusive thrombus in response to the stenosis and crush injury. In contrast, there was no occlusive thrombus formation in the carotid artery of the Nbeal2−/− mouse (n = 7, bar shows ± standard error of the mean [SEM]; P < .0001). (B) WT mice formed occlusive thrombi at an average time of 330 seconds after deendothelization of the stenosed vessel. Nbeal2−/− mice were unable to form occlusive thrombi in vivo (n = 7, bar shows ± SEM; P < .0005). (C) Representative macroscopic images of the gross anatomy of an excised carotid artery from WT and Nbeal2−/− mice. The WT carotid features a prominent clot (arrows) within the longitudinal arterial lumen (outlined) suggesting the presence of an occlusion. There is a clear absence of occlusive matter within the lumen of the Nbeal2 carotid (outlined). (D) Carstairs histological staining revealed that the WT thrombi were primarily composed of platelets (blue material in the lumen).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/14/10.1182_bloodadvances.2020002117/4/m_advancesadv2020002117f4.png?Expires=1767899350&Signature=Sq67NgMnF9I6mnTVkWZh6K-D0VSgfFltvKYP8birLSY-ZFLgTyAZ-Jh5f6gPh-Ui8-qQvVqHQPKfL~qG-I5qr2a0J1WCZe4ZKexqAWyKyUis3mQ6sQXoo3FgCJLwBpe8MQtJcyE9PbOoVexkljIaLAtPFkvyFXz8DBtZfPR7LXIG~~xQLvDGzgYgm3j1-930kd3hX~ENsJPvQk9NnXMh0c4Heb00EDGAt1yWYTrAvEAHeTsDVF2192KorlW8V9qxPAt1flnL7nY4lZLZmyqaJ9GjA49cOGuMKUWleB86Kl66zUGMyVjCTlLcK42sV1Zpd6NVswSy~k~qLZDhpWXteg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
An absence of platelet VWF prevents occlusive thrombus formation in the modified Folts arterial stenosis model. (A) Mean blood flow through the carotid artery was reduced to 50% by making stenosis and subsequently crush injury was induced. WT mice consistently formed an occlusive thrombus in response to the stenosis and crush injury. In contrast, there was no occlusive thrombus formation in the carotid artery of the Nbeal2−/− mouse (n = 7, bar shows ± standard error of the mean [SEM]; P < .0001). (B) WT mice formed occlusive thrombi at an average time of 330 seconds after deendothelization of the stenosed vessel. Nbeal2−/− mice were unable to form occlusive thrombi in vivo (n = 7, bar shows ± SEM; P < .0005). (C) Representative macroscopic images of the gross anatomy of an excised carotid artery from WT and Nbeal2−/− mice. The WT carotid features a prominent clot (arrows) within the longitudinal arterial lumen (outlined) suggesting the presence of an occlusion. There is a clear absence of occlusive matter within the lumen of the Nbeal2 carotid (outlined). (D) Carstairs histological staining revealed that the WT thrombi were primarily composed of platelets (blue material in the lumen).
An absence of platelet VWF prevents occlusive thrombus formation in the modified Folts arterial stenosis model. (A) Mean blood flow through the carotid artery was reduced to 50% by making stenosis and subsequently crush injury was induced. WT mice consistently formed an occlusive thrombus in response to the stenosis and crush injury. In contrast, there was no occlusive thrombus formation in the carotid artery of the Nbeal2−/− mouse (n = 7, bar shows ± standard error of the mean [SEM]; P < .0001). (B) WT mice formed occlusive thrombi at an average time of 330 seconds after deendothelization of the stenosed vessel. Nbeal2−/− mice were unable to form occlusive thrombi in vivo (n = 7, bar shows ± SEM; P < .0005). (C) Representative macroscopic images of the gross anatomy of an excised carotid artery from WT and Nbeal2−/− mice. The WT carotid features a prominent clot (arrows) within the longitudinal arterial lumen (outlined) suggesting the presence of an occlusion. There is a clear absence of occlusive matter within the lumen of the Nbeal2 carotid (outlined). (D) Carstairs histological staining revealed that the WT thrombi were primarily composed of platelets (blue material in the lumen).
Histological examination using Carstairs stain revealed that the thrombi in the occluded arteries of WT mice were platelet rich, whereas histology and macroscopic imaging showed an absence of thrombi at the vessel injury site in Nbeal2−/− mice (Figure 4C-D). These findings indicate that VWF from platelet α-granules is necessary for occlusive thrombus formation in a stenosed artery.
Platelet VWF is critical for the RPA phase and subsequent vessel occlusion
Mehrabadi et al45 modeled high-shear thrombosis with 2 phases: lag and RPA. We applied their thrombosis growth model for microfluidics and mouse carotid artery, comparing occlusion times. Microfluidic simulations of an 82-μm-height channel predicted a lag phase time of 138 seconds, followed by an RPA phase of 109 seconds, resulting in a total occlusion time of 247 seconds, which is comparable to the experimental result (326 ± 25 seconds). Nbeal2−/− mouse whole blood did not advance past the lag phase, and no RPA was observed. However, when new exogenous WT mouse platelets were perfused through to the same device channel containing Nbeal2−/− adherent platelets, the RPA phase occurred almost immediately (bypassing its own WT lag phase). Subtracting the lag time of the Nbeal2−/− platelets from the RPA phase of the WT platelets showed an occlusion time of 170 ± 57 seconds, which was comparable to the predicted value for RPA occlusion of 138 seconds (Figure 5).

Comparison of simulated occlusion times vs actual experimental values. (A) In vitro microfluidics. (B) In vivo mouse carotid artery modified Folts model.
Comparison of simulated occlusion times vs actual experimental values. (A) In vitro microfluidics. (B) In vivo mouse carotid artery modified Folts model.
In simulations of a mouse carotid artery stenosed to 60% of its diameter, the modeled occlusion time was calculated as 305 seconds, which was similar to the results of in vivo experiments in WT mice, which exhibited a mean occlusion time of 352 seconds (Figure 5).
Discussion
We investigated the role of platelet VWF in high-shear occlusive thrombus formation using a combination of CFDs: high-shear in vitro microfluidics and in vivo stenosis model of arterial thrombosis. Many studies of platelets investigate attachment at physiological shear rates. Here, high-shear thrombus formation is different from physiological platelet attachment because of the increased shear rates and high hemodynamic forces caused by an atherosclerotic stenosis in a critical artery. As thrombus formation progresses at these stenoses, the diameter of the throat narrows, further increasing the shear rates. Flow occlusion and occlusion time are relevant end points that correspond to acute coronary syndrome. Using models that closely mimic the geometries of stenotic arteries, we recapitulated the time to occlusive thrombosis and correlated and compared in silico simulations with in vitro and in vivo experiments.
The microfluidic channel and mouse carotid artery were modeled and simulated by calculating the shear rates through their respective geometries. Using the predicted shear rates, occlusion times were determined with a 2-phase model.45 Comparisons with WT experimental data showed that the model could predict shear rates and occlusion times for a ∼60% stenosis where normal plasma and platelet VWF was present. Knowing that our model had good predictivity for normal WT whole blood, we went on to determine the individual contributions of VWF in the plasma and platelet compartments of whole blood in Nbeal2−/− mice that do not have α-granules and consequently no platelet VWF.12 Recapitulating the CFDs, we found that Nbeal2−/− mice blood could not form occlusive thrombi in microfluidic chambers as compared with WT blood. In the microfluidic assays, Nbeal2−/− platelets only minimally adhered to collagen under high shear during the lag phase. No RPA or occlusion in the microchannels was observed with Nbeal2−/− mouse blood. Amelioration of the RPA phase was observed when WT platelets with normal platelet VWF were perfused through the same microfluidic channel. This effect was sufficient to restore normal RPA and localized occlusive thrombus formation as a Rescue study. We were able to validate that Nbeal2−/− platelets also showed impaired thrombus formation in vivo using a modified Folts model with high shear compared with WT controls. By correlating shear rates from computational fluid dynamics modeling, this work could be used to predict the thrombogenic potential of implanted blood contacting medical devices, such as artificial circulation and heart valves, that are prone to causing occlusive arterial thrombosis.49
Although platelet VWF is of central importance for the formation of shear-resistant occlusive thrombosis under these pathological experimental shear rates, it cannot be ruled out that other α-granule adhesive proteins and coagulation factors may also contribute to this process, although the contribution of these proteins at pathological shear may be less significant. Future studies in transgenic mice in which platelet VWF is ablated are needed to further dissect this mechanism.
Our Nbeal2−/− mice did not contain platelet VWF, and their plasma VWF levels were normal. In WT platelets, the VWF inside α-granules was 50-fold higher than the concentration of plasma VWF27 and was capable of mediating platelet adhesion >3000 s−1. 29 As platelets become activated and release their α-granule contents, the localized release of high concentrations of ULM VWF is delivered directly to the thrombus cap. Although Nbeal2−/− mice are reported to have mild thrombocytopenia,12,24 previous reports have shown that severe platelet count reduction, including the thrombocytopenia phenotype observed in Nbeal2−/− mice, have no major effect on hemostasis and thrombosis.23,24,50 Therefore, it is highly unlikely that our thrombotic defect was related to a reduced platelet count in Nbeal2−/− mice.
In in vivo experiments using the common carotid artery, shear rates at the stenosis where there was a 50% reduction in blood flow were estimated to be >4600 s−1. Under these conditions, similar to human thrombus formation, mouse platelet thrombus formation at this shear is assumed to be VWF dependent. Though platelet activation and coagulation are mutually dependent processes, we speculate that in vivo platelet VWF in occlusive thrombosis is coagulation pathway independent, most likely because of the rapid transport away of coagulation factors and the diffusion of assembled coagulation factors at the site of vascular damage by increased pathological shear rates. We used heparinized (15 USP units/mL) whole blood in vitro, so that we could investigate specific platelet-matrix/platelet-platelet interactions without coagulation while transporting the blood. In the in vitro experiments, in the absence of coagulation, we observe RPA (and occlusion) only when platelet (and plasma) VWF was present. Occlusion did not occur in the absence of coagulation and platelet VWF. Although the interpretation of the in vitro results cannot be directly compared against in vivo results where coagulation can occur, the study still demonstrates that platelet VWF is required for occlusive thrombosis to occur in both models. These findings demonstrate that α-granule–derived VWF, released after activation, contributes to thrombus growth independent of the plasma VWF levels that would aid initial platelet adhesion before activation.
In conclusion, the results of this study show the importance of shear-induced platelet VWF release that creates an occlusive thrombus. Theoretical predictions combined with in vitro and in vivo experiments isolated the necessary role of α-granules for large thrombus formation. Current antithrombotic therapeutics are unable to distinguish between physiological and pathological shear rates without affecting hemostasis. The requirement of platelet VWF to bind platelets under high-shear conditions, like those in stenotic arteries, makes it a potential novel target for pharmacological intervention to prevent pathological thrombus formation in myocardial infarction or stroke.
Original data are available by e-mail request to the corresponding author.
Authorship
Contribution: D.A.K. and D.N.K designed the study; D.A.K. and K.J.A. performed the experiments; and all authors wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David N. Ku, Georgia Institute of Technology, G. W. Woodruff School of Mechanical Engineering, 315 Ferst Dr NW, IBB 2307, Atlanta, GA 30332; e-mail: david.ku@me.gatech.edu.



