Key Points
Leukemic evolution of PV and ET harbored distinct molecular profiles associated with different time to transformation.
Additional mutations in the chronic phase were associated with a higher risk of leukemic evolution.

Abstract
Among myeloproliferative neoplasms, polycythemia vera (PV) and essential thrombocythemia (ET) are the 2 entities associated with the most chronic disease course. Leukemic evolution occurs rarely but has a grim prognosis. The interval between diagnosis and leukemic evolution is highly variable, from a few years to >20 years. We performed a molecular evaluation of 49 leukemic transformations of PV and ET by targeted next-generation sequencing. Using a hierarchical classification, we identified 3 molecular groups associated with a distinct time to leukemic transformation. Short-term transformations were mostly characterized by a complex molecular landscape and mutations in IDH1/2, RUNX1, and U2AF1 genes, whereas long-term transformations were associated with mutations in TP53, NRAS, and BCORL1 genes. Studying paired samples from chronic phase and transformation, we detected some mutations already present during the chronic phase, either with a significant allele burden (short-term transformation) or with a very low allele burden (especially TP53 mutations). However, other mutations were not detected even 1 year before leukemic transformation. Our results suggest that the leukemic transformation of PV and ET may be driven by distinct time-dependent molecular mechanisms.
Introduction
Myeloproliferative neoplasms (MPNs) are acquired clonal disorders characterized by the proliferation and accumulation of mature blood cells. Classic Philadelphia-negative MPNs include polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). PV and ET are the most indolent diseases, with a median overall survival (OS) of >12 and 15 years, respectively.1 The prognosis of these diseases is related to 2 major complications: thrombosis in the short and midterm and hematological transformations in the long term. Among the hematological transformations, evolution to secondary acute myeloid leukemia (AML) is associated with a poor prognosis, with a median OS below 6 months.2-4 The incidence of leukemic transformation of PV and ET lies between 2% and 5% at 15 years.5,6 The interval between diagnosis and leukemic progression is highly variable from 1 patient to another, from a few months or years to more than 20 years, and the identification of high risk-patients remains difficult.2,3,7,8
Previously reported risk factors for leukemic transformation in PV and ET include older age (>60, 65, or 70 years, according to studies); anemia, leukocytosis, thrombocytopenia, and thrombocytosis >1000 ×109/L; or abnormal karyotype.4-6,9,10 In ET, triple-negative patients seem to have a slightly higher risk of leukemic evolution than those with classic driver mutations.11 Some additional mutations at diagnosis have also been associated with leukemic evolution. Recently, Grinfeld et al12 proposed a molecular prognostic classification in 8 classes for all MPN, where both TP53 mutations and mutations in the chromatin/spliceosome (including ASXL1, EZH2, and SRSF2) were associated with a higher risk of leukemic evolution. In another recent study, a high-risk group included patients with PV or ET with mutations of SRSF2 for PV and SRSF2, SF3B1, U2AF1, and TP53 for ET.13
Leukemic transformations are characterized by mutations in TP53, ASXL1, RUNX1, EZH2, IDH1/2, and SH2B3.14-17 These mutations may be acquired only at the time of leukemic transformation or are already detectable during the chronic phase, although at a very low level.18,19 The driver mutation JAK2V617F can also disappear during leukemic transformation and thus reflect clonal evolution.20 However, the genetic mechanisms of this evolution are not completely clarified.
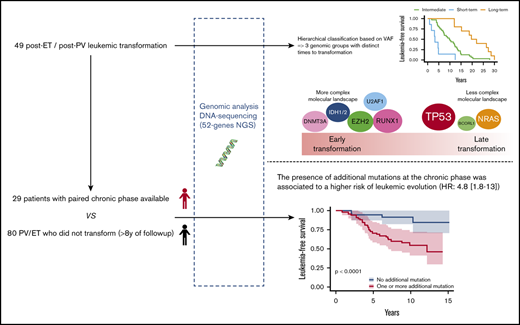
We studied a multicentric cohort of 49 patients with post-PV/ET AML at the time of leukemic transformation and at the chronic phase by next-generation sequencing (NGS). We identified a molecular classification of leukemic transformation associated with different times to transformation.
Material and methods
Patients and samples
Patients from 4 French university hospitals (Angers, Brest, Poitiers, and Rennes) were included if they had PV or ET with leukemic evolution and DNA samples available at the time of leukemic evolution, which was defined by marrow or peripheral blood blast counts ≥20%. Patients treated with busulfan or 32P were excluded. A total of 49 patients were included in the study. Diagnoses were performed according to World Health Organization 2001 (WHO 2001) or WHO 2008 criteria. Molecular analyses were performed at the time of leukemic transformation, and a paired diagnostic or chronic phase was also analyzed, if available. Patients in the chronic phase were defined as those without progression to myelofibrosis or myelodysplasia and without cytopenia or circulating blasts. If the diagnostic sample was not available, the oldest sample in chronic phase was studied. For leukemic stage, the sample with the highest blast infiltration was studied. In 7 patients, additional samples obtained during follow-up, before leukemic transformation, were also analyzed. To investigate the prognosis of additional mutations during chronic phase, a control group of 80 patients (37 PV and 43 ET) who did not experience transformation with at least 8 years of follow-up were studied (details in supplemental Methods). This study was registered with the CNIL (Commission nationale de l'informatique et des libertés, French Data Protection Authority, n° 2016-070) and samples were provided from the “Hemopathies Malignes” biobank of Angers University Hospitals, which was approved by the CPP (“Comité de Protection des Personnes,” Institutional Review Board) of Angers Ouest II.
NGS
NGS for all coding exons of 52 genes was performed by using a HaloPlexHS custom panel (Agilent Technology, Santa Clara, CA). Library preparation and sequencing with NextSeq500 (Illumina, San Diego, CA) were performed according to the manufacturer’s instructions (details in supplemental Data). Nonsynonymous coding variants were retained when variant allele frequency (VAF) was higher than 2%, and coverage of the called regions was set to 50. Somatic mutations were then classified according to their putative impact in 3 groups: deleterious variant, probable deleterious variant, and variant of undetermined significance, according to the recommendations of the American Society of Genetics and the guidelines for interpretation of somatic variants21,22 (supplemental Table 1).
Statistics
Statistical analysis was performed with R software (version 3.5.1, www.R-project.org, Vienna, Austria). Subjects’ characteristics were reported as number and percentage for qualitative variables and as the mean ± standard deviation or median [interquartile range (IQR)], as appropriate, for continuous variables. Data were compared by using Fisher’s exact test for categorical variables and the Mann-Whitney U test or Kruskal-Wallis test for continuous variables. The violin plot was used to compare the distribution of allele burden in mutations, with the boxes indicating the 75th percentile (top horizontal line), median (black bold horizontal line), and the 25th (bottom horizontal line) percentiles of the distribution, and a rotated kernel density plot surrounding the box (shaded area) on each side. The sparse partial least-squares discriminant analysis (sPLS) method was used to predict time to AML progression and to evaluate which additional mutation enables class-distinguishing information.23,24 Then a hierarchical clustering classification was performed on the 2 major axes of the sPLS, according to Euclidean distance.25,26 Survival analyses were performed with multivariate Cox modeling. The Kaplan-Meier estimate for graphic representation of the univariate approach was used. The specific causal effect of additional mutations during the chronic phase on leukemic transformation was studied by using a weighting-based propensity score (inverse probability weighting). The point of sampling was time 0. Kaplan-Meier curves and Cox models were then used, taking this weighting into account.
Statistical analyses were performed considering all additional mutations, except for the sensitivity analysis of sPLS and prognostic evaluation of additional mutations at the chronic phase, where only deleterious and probable deleterious mutations were considered.
Results
Patient characteristics at the time of diagnosis and at leukemic transformation
The clinical and biological characteristics of the 49 patients with MPN (24 with PV and 25 with ET; supplemental Figure 1) are summarized in Table 1. There was a slight male predominance (61% of patients), especially in patients with PV (75% of patients). The median age at the time of MPN diagnosis was 63 years, and the time to leukemic progression ranged from 1 to 30 years (median, 12 years). The distribution of driver mutations was as expected for patients with PV or ET.27 Of note, all CALR-mutated ET showed late transformation beyond 12 years of follow-up. At leukemic transformation, the median age was 74 years (range, 38-88). The blood counts (median; range) at leukemic transformation showed anemia in 87% (8.6 g/dL; 4.2-14.1), thrombocytopenia in 69% (80 ×109/L; 9-630), and neutropenia in 27% (1.85 ×109/L; 0.08-38) of cases. A high leukocyte count was observed in 39% of cases of leukemic transformation (7.7 ×109/L; 0.75-101). Karyotype was normal in 37% (13 of 35) and complex in 26% (9 of 35) of cases.
Mutational landscape of post PV/ET-AML
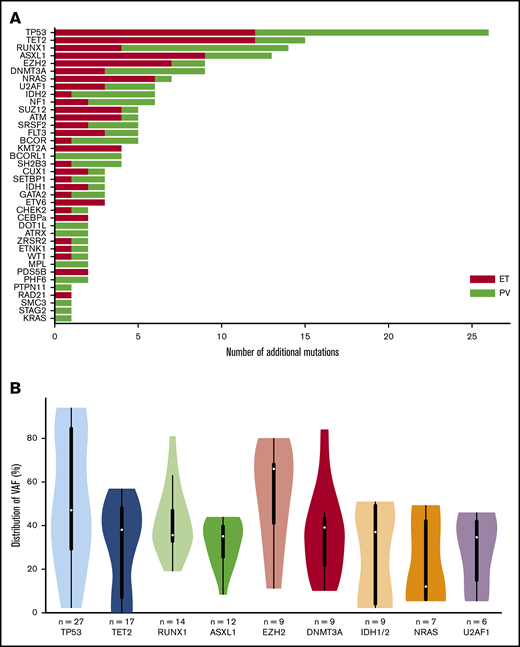
In the 49 post-PV/ET AML, we detected a total of 191 additional mutations (ie, not including driver mutations). All patients carried at least 1 additional mutation. The median number of additional mutations was 4 (range, 1-8). The most frequently mutated genes were TP53, TET2, RUNX1, ASXL1, and EZH2 (Figure 1A). No difference was found in mutated genes between patients after PV or ET. VAFs are plotted in Figure 1B. The median VAF of TP53 mutations was 47%, 37% of which had a VAF above 60%, indicating loss of heterozygosity. Five patients harbored 2 TP53 mutations, and for 3 of them, the 2 mutations were close together, thus corresponding to different clones or separate alleles in the same clone. Five of the 9 mutations of EZH2 had an allele burden >50%.
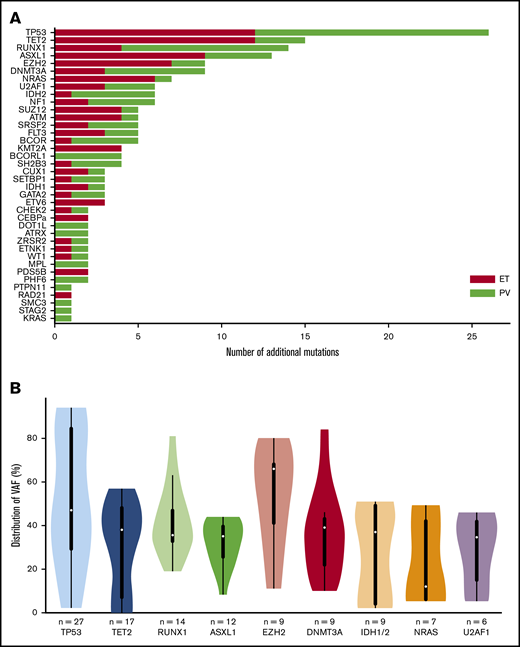
Genes mutated at the time of leukemic evolution of MPNs. (A) The total number of mutations is represented. Mutations in post-PV AML are in red, and those in post-ET AML are in green. (B) Violin plots of the VAF distribution of genes with more than 6 mutations. IDH1 and IDH2 have been summed for this figure.
Genes mutated at the time of leukemic evolution of MPNs. (A) The total number of mutations is represented. Mutations in post-PV AML are in red, and those in post-ET AML are in green. (B) Violin plots of the VAF distribution of genes with more than 6 mutations. IDH1 and IDH2 have been summed for this figure.
Because time to transformation varied significantly between patients, we wondered if this could be related to the genes affected by additional mutations. For this purpose, to predict the time to leukemic progression, we performed an sPLS analysis comparing all additional mutations. Each mutation was considered as a quantitative variable based on its allele burden. This analysis allowed for clustering of patients according to the time to leukemic transformation. In particular, mutations of TP53, BCORL1, and NRAS were associated with late transformations, whereas mutations of U2AF1, IDH1, IDH2, EZH2, and DNMT3A were associated with an earlier time to transformation (supplemental Figure 2). A sensitivity analysis considering only deleterious variants and probable deleterious variants found similar results (supplemental Figure 3).
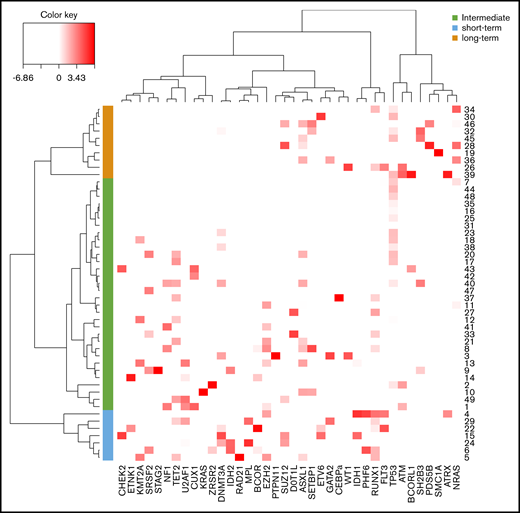
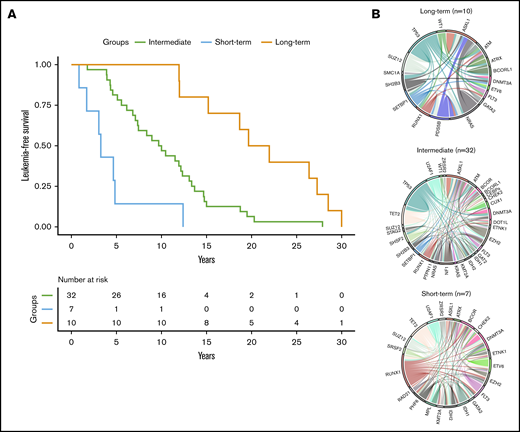
Using a hierarchical classification on the sPLS data, we identified 3 groups of patients associated with different time to leukemic transformation (Figures 2 and 3A). The median time to transformation was 3, 10, and 21 years for the short-term, intermediate, and long-term groups, respectively (Table 2). For the short-term group, all except 1 transformation occurred within 5 years of diagnosis, whereas for the long-term group, no transformation occurred within 12 years of follow-up.
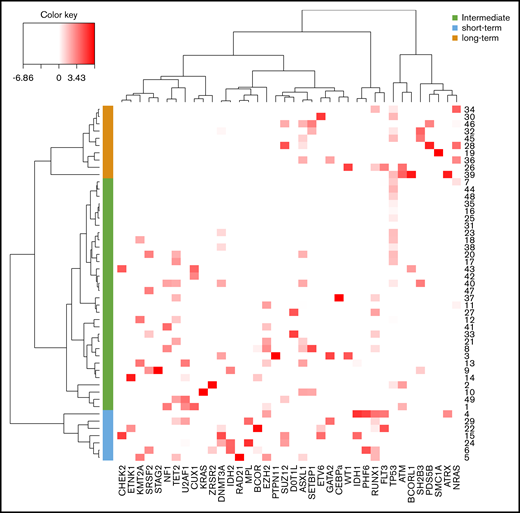
Classification of post-PV/ET AML according to their mutational landscape. Heat map based on the Euclidean distance, displaying the VAF of additional mutations. Genes are listed on the x-axis, and each row corresponds to a patient. The color scale represents the scaled abundance of each variable and is proportional to the allele burden (darker red represents higher allele burden). Three groups were identified for short-term (blue), long-term (orange), and intermediate (green) transformation.
Classification of post-PV/ET AML according to their mutational landscape. Heat map based on the Euclidean distance, displaying the VAF of additional mutations. Genes are listed on the x-axis, and each row corresponds to a patient. The color scale represents the scaled abundance of each variable and is proportional to the allele burden (darker red represents higher allele burden). Three groups were identified for short-term (blue), long-term (orange), and intermediate (green) transformation.
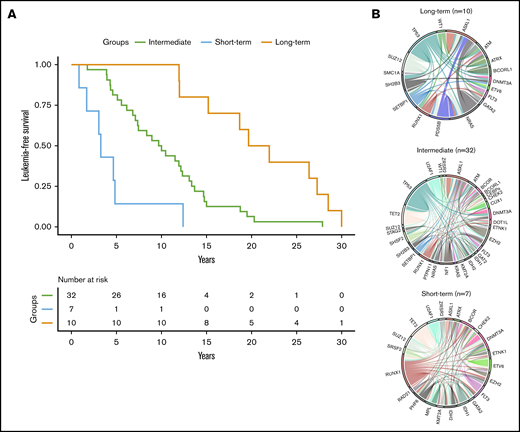
Time to leukemic transformation and the molecular landscape of the 3 groups of patients with post-PV/ET AML. (A) Kaplan-Meier curves for leukemia-free survival of the cohort, divided according to the length of time to leukemic transformation. (B) Circos plots show the molecular complexity of the 3 groups.
Time to leukemic transformation and the molecular landscape of the 3 groups of patients with post-PV/ET AML. (A) Kaplan-Meier curves for leukemia-free survival of the cohort, divided according to the length of time to leukemic transformation. (B) Circos plots show the molecular complexity of the 3 groups.
The 3 groups of leukemic transformation exhibited distinct molecular landscapes as shown in the Circos plots (Figure 3B; http://circos.ca/intro/genomic_data/). Patients with short-term transformation exhibited a complex molecular landscape with a median of 7 additional mutations. In this group, mutations occurred more specifically in RUNX1, IDH1/2, U2AF1, and TET2 genes (Table 2). On the other hand, patients with a long-term transformation presented, on the contrary, a less complex molecular landscape with preferential mutations in the TP53 and NRAS genes. Of note, no TP53 mutation was found in short-term transformation, whereas no mutations of IDH1/2, EZH2, U2AF1, and TET2 were found in late transformations. Mutations of ASXL1 were equally distributed in all 3 groups.
Mutational landscape of matched diagnosis/chronic phase
Matched samples obtained at diagnosis or during the chronic phase were studied for 29 of 49 patients. In detail, DNA from diagnosis samples was available for 21 patients. For the 8 other patients, we analyzed a sample obtained during the chronic phase of ET or PV that met the criteria described in “Materials and methods.” The driver mutation JAK2V617F disappeared at leukemic transformation in 8 of 23 cases (35%; supplemental Table 2). In the 29 patients studied in the diagnosis/chronic phase, 149 mutations were detected at leukemic transformation, including 83 (56%) detected at diagnosis (supplemental Figure 4). In addition, 14 mutations were also present at diagnosis, but with a low allele burden, and have been identified retrospectively by examination of the BAM sequencing files. Mutations of the IDH1/2, TET2, DNMT3A, and splicing (SRSF2, U2AF1, and ZRSR2) genes were frequently present at diagnosis (68% of cases, supplemental Table 3; supplemental Figure 4), whereas mutations in NRAS and RUNX1 were rarely detected at diagnosis. Half of the TP53 mutations were already present in the diagnosis/chronic phase, but at low levels: all had a VAF <5% except for 1 patient with ET who had a VAF of 26% at diagnosis. The patient was treated with pipobroman during the chronic phase but the ET evolved into acute leukemia over 8 years, with a marked increase in TP53 allele burden (76%). The evolution of allele burdens between diagnosis/chronic phases and leukemic transformations showed a significant increase in TP53, RUNX1, BCOR, EZH2, and ASXL1 mutations (supplemental Figure 5).
Finally, in 7 patients, we analyzed 9 intermediate samples between diagnosis and leukemic transformation, to track the onset or increase in mutations (supplemental Table 4). In 5 of those patients, several mutations that appeared at leukemic transformation were not detectable 1 to 2 years earlier, in particular in the TP53 (3 variants) and RUNX1 (2 variants) genes. In another patient, the acquisition of 3 mutations in ASXL1, EZH2, and NRAS was detectable 1 year before evolution to acute leukemia (supplemental Figure 6). At this time, the patient had unexplained anemia not related to treatment, and bone marrow examination was not in favor of myelodysplastic syndrome.
The evolution of the allele burden of mutations between the chronic and leukemic phases provided information on the acquisition order in some cases (supplemental Figure 7). TP53 mutations were late events mostly arising outside the driver mutation (7 of 9; 78% of cases). CALR driver mutations were encountered in the founder clone, whereas JAK2V617F could be early or secondary events (supplemental Figure 7).
A patient had 2 TP53 mutations at the time of diagnosis with very low allele burdens (1.4% and 3%), with one of them increasing during 11 years of follow-up to 13%, whereas the other remained stable at 3%. At leukemic transformation 1 year later, the low-level mutation had disappeared, whereas the second one had increased dramatically to 84%.
Additional mutations during chronic phase and risk of leukemic transformation
We then studied the impact of additional mutations during the chronic phase on the risk of leukemic transformation. For this purpose, we performed a case-control analysis with 29 patients with post-PV or -ET leukemia with a chronic sample, and 80 control patients (37 PV and 43 ET) who did not transform to acute leukemia during at least 8 years of follow-up (range, 8-29). For this analysis, only deleterious and probable deleterious mutations were considered, as the prognostic impact of variants of unknown significance is unclear, and they may in fact be rare polymorphisms.
In the control cohort of stable patients, additional mutations were detected in 28 of 80 cases (35%), and TET2 was the most mutated gene, with 11 of 80 patients having at least 1 mutation (14%).
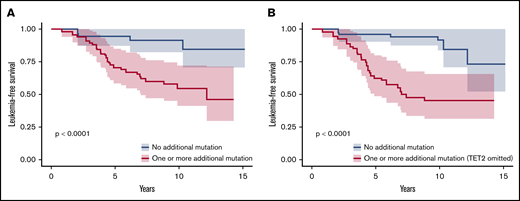
We adjusted data on age at the time of sampling, sex, type of disease (PV or ET) and driver mutation by using a weighting propensity score to assess causality between additional mutations and leukemic transformation. The presence of at least 1 additional mutation during chronic phase was associated with a higher risk of leukemic evolution (Figure 4A; P < .0001). In multivariate analysis, both age at diagnosis (hazard ratio [HR], 1.07; 95% confidence interval [CI], 1.03-1.12; P = .0004) and the presence of an additional mutation (HR, 4.8; 95% CI, 1.8-13; P = .002) were associated with a risk of leukemic transformation (supplemental Table 5). After exclusion of TET2 mutations from the analysis, the association between an additional mutation and the risk of leukemic evolution was stronger (HR, 7.3; 95% CI, 2.9-18; P < .0001; Figure 4B).
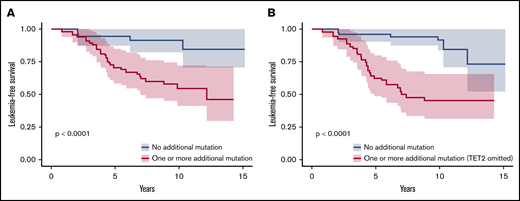
Leukemia-free survival of patients during the chronic phase according to the presence of additional mutations. Kaplan-Meier curve of leukemia-free survival for patients in the chronic phase (n = 109; 80 patients from the control group and 29 patients evolving to leukemic transformation) after propensity score adjustment according to the presence of at least 1 additional mutation, considering all additional mutations (A) or all additional mutations except in TET2 (B).
Leukemia-free survival of patients during the chronic phase according to the presence of additional mutations. Kaplan-Meier curve of leukemia-free survival for patients in the chronic phase (n = 109; 80 patients from the control group and 29 patients evolving to leukemic transformation) after propensity score adjustment according to the presence of at least 1 additional mutation, considering all additional mutations (A) or all additional mutations except in TET2 (B).
Prognosis of post-PV/ET AML
The median OS of leukemic transformation for the study cohort was 8 months. For patients treated with best supportive care (BSC), nonintensive therapy, and hematopoietic stem cell transplantation (SCT), the 18-month OS was 9%, 15%, and 50%, respectively (P = .015; supplemental Figure 8). Patients who received intensive therapy without SCT had a prognosis comparable to that of patients treated with BSC. The median OS between the 3 transformation time groups was 4, 8, and 19 months for the intermediate, long-term, and short-term groups, respectively (P = .24, supplemental Figure 9). Finally, we examined the prognostic impact of additional genes mutated in at least 10 patients at leukemic transformation (TET2, ASXL1, RUNX1, and TP53). We observed decreased survival in patients with TP53 mutations vs wild-type, with a 12-month OS of 18% vs 48% (P = .05; supplemental Figure 10).
Discussion
We studied 49 patients with PV or ET that evolved into acute myeloid leukemia, and we analyzed the molecular landscape of both leukemic transformation and matched diagnosis or chronic phase, if the information was available.
In the present study, TP53 was the most mutated gene at leukemic transformation, with 45% of patients bearing the mutation. TP53 mutations have been previously described in post-MPN acute leukemia, and the involvement of p53 inactivation in leukemogenesis has been supported in a mouse model.15,18 The rate of TP53 mutations found in this study is higher than in previous studies (16% to 34%)16,17,28 and may have been the result of our studying patients with PV or ET and not those with PMF. Indeed, Courtier et al found TP53 mutations in 42% of post-PV/ET AML and in 0 of 7 post-PMF AML.16 We focused our study in PV and ET because leukemic transformation is a rare, usually late complication and has been less documented. We also assumed that myelofibrosis is a molecularly more complex disease and that the mechanisms of leukemic transformation would be different. Prefibrotic PMF, integrated in the WHO 2016 classification, is associated with a higher risk of leukemic transformation.29 All available bone marrow biopsies were reviewed (18 of 25 ET). Typical aspects of ET were found in 14 patients and features consistent with prefibrotic myelofibrosis were found in 4 patients. However, 2 of the 4 patients lacked a WHO 2016 minor criteria, as previously described, and would be diagnosed as unclassifiable MPN.30 We found no differences between these 4 patients and patients with ET in the number (median; range) of additional mutations at the chronic phase (2; 1-2 vs 2; 1-7; P = .33) or time to transformation (10; 7-12 vs 11; 1-29; P = .83).
In this study, we proposed a classification based on the mutations at leukemic transformation that considered not only the presence or absence of mutations, but also the allele burden. We assumed that VAF of additional mutations better reflects the oncogenic role of each mutation in the leukemic transformation process. Indeed, mutations with a low allele burden would rather be passengers, whereas mutations with a high allele burden would rather be drivers of leukemic transformation. Nevertheless, the only allele burden could not be sufficient to characterize a driver mutation.31 Using this methodology, we identified 3 distinct genomic groups associated with distinct transformation times.
For short-term transformations, the molecular landscape was mostly complex with a combination of mutations in genes involved in different functions (epigenetic, signaling, splicing, or transcription factors). Part of these mutations had been present since diagnosis, which is consistent with more complex molecular diseases. This observation fits with previous reports that sought to identify patients with a molecular disease.12,13,19,32 All 7 patients of the short-term group exhibited at least 1 mutation of the chromatin/spliceosome category associated with a higher risk of leukemic evolution in the classification proposed by Grinfeld et al.12 In addition to the additional mutations that were present at the time of diagnosis, other mutations were acquired at leukemic transformation, suggesting a role of these secondary events as triggers of the transformation. Indeed, RUNX1 mutations are often acquired at leukemic transformation and are involved in leukemogenesis by inducing a myeloid differentiation block.33 Notably, no CALR-mutated MPN had a short-term transformation that could be related to the less complex molecular landscape of these patients34 and could reflect a lower genetic instability than in JAK2V617F MPN.35
For the intermediate and long-term groups, the molecular landscape was less complex, with specific mutations in the TP53, ATM, NRAS, or BCORL1 genes. These mutations could have been present since diagnosis, often at a low level, or they could appear during leukemic transformation. Moreover, in some cases, the mutations were not detectable even a few months before leukemic transformation within the sensitivity limits of our technology, which ranges from 0.1% to 0.5%. The presence of TP53 mutations at a low level in the chronic phase of MPN has been described.18,19,36 The association between TP53 mutations and long-term transformation could be related to the necessary inactivation of the second allele of TP53 for leukemic transformation.18 Indeed, we found an allele burden higher than 50% or a second mutation in most of the of leukemic transformations with the TP53 mutation. On the basis of therapy-related AML, the putative role of the cytoreductive treatment in the selection of the TP53 mutated clone is still discussed.37 To date, there is no sufficient argument for this hypothesis in post-MPN AML. In a previous study, TP53 mutations were found to be associated with >4 years treatment with hydroxycarbamide, but the association was lost after age adjustment.36 Our finding of TP53 mutations associated with later transformation could support a role of long-term treatment, but it could also be related to other mechanisms, such as disease-associated genetic instability or altered bone marrow niche.35,38 The association between pipobroman exposure and late leukemic transformation has been shown.6,39 In our study, 16 patients had been treated for more than 12 months with pipobroman, mostly as a second- or third-line therapy, and those patients more frequently harbored a TP53 mutation at leukemic transformation (75% vs 31%; P = .005; supplemental Figure 11).
We then investigated the impact of additional mutations during the chronic phase on the risk of leukemic evolution with the analysis of a control group of patients with stable PV or ET with a long follow-up. We found that the presence of at least 1 additional mutation during the chronic phase was associated with a higher risk of leukemic transformation. This association was higher when TET2 mutations were omitted. Mutations in the TET2 gene are the most frequently encountered in PV and ET and have shown no prognostic impact in previous studies.19,40,41 Those studies have reported such an association between additional mutations and leukemic evolution in a smaller number of transformation cases, because it is a rare and late event in PV and ET.19,42,43 Recently, a molecular high-risk signature has been developed in PV (SRSF2 mutations) and ET (SRSF2, SF3B1, U2AF1, and TP53 mutations).13 In our study, a high-risk mutation during the chronic phase was associated with a higher risk of leukemic evolution (HR, 6.9; 95% CI, 3.1-15.1; P < .0001; supplemental Figure 12).
The leukemic evolution of myeloproliferative neoplasms is associated with a poor prognosis. In this study, we found a median OS of 8 months, consistent with previous studies.2-4,28,44,45 As previously described, the benefit of intensive therapy seems restricted to patient who undergo allogenic SCT.3,28 Nevertheless, the survival of transplant recipients was also very poor, suggesting that allogenic SCT should be considered before transformation in high-risk patients.28,45 In our study, nonintensive therapy with low-dose aracytine or azacytidine is associated with a better outcome than BSC or intensive therapy without transplantation. Several mutations associated with an adverse prognosis at the leukemic transformation have been described in particular in the TP53, RUNX1, and SRSF2 genes.17,28,44,46 The slightly better prognosis of patients with a short-term transformation could be caused by the absence of TP53 mutations, a younger age at the time of leukemic evolution, and the inferior use of supportive care.
In summary, our results suggest that the leukemic evolution of PV and ET involves different mutations according to time to transformation. Both larger cohorts and functional studies are needed to further clarify molecular mechanisms associated with short-term and long-term transformation. Obviously, analysis of the clonal architecture of leukemic transformation of MPN may improve understanding of the driver of leukemic evolution.
Genomics data are available upon request from the corresponding author (damien.luquepaz@chu-angers.fr).
Acknowledgments
The authors thank Gérard Socié for helpful scientific discussions and critical reading of the manuscript; the local tumour biobanks of Brest and Angers and the French Clinical and Biological Network of Myeloproliferative Neoplasms (FIMBANK) for providing high-quality samples; and the French Intergroup of Myeloproliferative Neoplasms (FIM) for scientific discussions. This work was realized in the context of the 3I-Impact project (The University of Angers and the University Hospital of Angers joint program) and of the HUGO network for NGS in MPN (Bourse Espoir HUGO 2017).
This work was supported by grants from the University Hospital of Angers (AO Interne 2016) (D.L.P.) and the League Against Cancer (Ligue Contre le Cancer 49, appel d’offre régional 2018) (D.L.P.).
Authorship
Contribution: D.L.P. and V.U. conceived and designed the study; D.L.P., R.J.-C., and A.C. collected the clinical data; O.B. supervised the biobanking; J.-M.C. conceived and supervised the biological clinical database of the FIMBANK project; D.L.P., R.J.-C., A.C., J.-C.C., L.C., C.P., M.R., A.M., and F.C. performed the NGS experiments and analyzed and interpreted the data; J.-C.I., F.B., E.C., M.-P.G.-H., C.O., S.T., N.I., and M.H.-B. cared for the patients; D.L.P., R.J.-C., J.R., and M.R. analyzed the data; M.-C.R. and I.Q.-R reviewed the bone-marrow biopsies; D.L.P., R.J.-C., and V.U. wrote the manuscript; and J.-C.I., C.O., Y.D., N.I., E.L., and M.H. revised it critically.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Damien Luque Paz, Laboratoire d’Hématologie, Institut de Biologie en Santé, CHU Angers, 4 Rue Larrey, 49933 Angers Cedex 9, France; e-mail: damien.luquepaz@chu-angers.fr.