Key Points
HuMkMPs target and program megakaryocytic differentiation of muHSPCs in vitro.
Intravenous administration of huMkMPs enables de novo platelet biogenesis in vivo.

Abstract
Platelet transfusions are used to treat idiopathic or drug-induced thrombocytopenia. Platelets are an expensive product in limited supply, with limited storage and distribution capabilities because they cannot be frozen. We have demonstrated that, in vitro, human megakaryocytic microparticles (huMkMPs) target human CD34+ hematopoietic stem and progenitor cells (huHSPCs) and induce their Mk differentiation and platelet biogenesis in the absence of thrombopoietin. In this study, we showed that, in vitro, huMkMPs can also target murine HSPCs (muHSPCs) to induce them to differentiate into megakaryocytes in the absence of thrombopoietin. Based on that, using wild-type BALB/c mice, we demonstrated that intravenously administering 2 × 106 huMkMPs triggered de novo murine platelet biogenesis to increase platelet levels up to 49% 16 hours after administration. huMkMPs also largely rescued low platelet levels in mice with induced thrombocytopenia 16 hours after administration by increasing platelet counts by 51%, compared with platelet counts in thrombocytopenic mice. Normalized on a tissue-mass basis, biodistribution experiments show that MkMPs localized largely to the bone marrow, lungs, and liver 24 hours after huMkMP administration. Beyond the bone marrow, CD41+ (megakaryocytes and Mk-progenitor) cells were frequent in lungs, spleen, and especially, liver. In the liver, infused huMKMPs colocalized with Mk progenitors and muHSPCs, thus suggesting that huMkMPs interact with muHSPCs in vivo to induce platelet biogenesis. Our data demonstrate the potential of huMkMPs, which can be stored frozen, to treat thrombocytopenias and serve as effective carriers for in vivo, target-specific cargo delivery to HSPCs.
Introduction
Megakaryocytes (Mks) are large, polyploid (≥8N) cells derived from hematopoietic stem and progenitor cells (HSPCs), contained within the CD34+-cell compartment, which, upon maturation induced by thrombopoietin (TPO) activity, shed pre- and proplatelets (PPTs) and platelet-like particles (PLPs) that mature quickly into platelets.1-3 Mks also shed CD41+CD42b+CD62P− Mk microparticles (MkMPs).4,5 Cell-derived microparticles/microvesicles (MPs/MVs) are submicrometer-size extracellular vesicles (EVs) that are emerging as potential therapeutic agents.6,7 It has been known since 2009 that MkMPs are the most abundant MPs in circulation and that they are distinct from platelet-derived MPs (PMPs).5
In transfusion medicine, platelets are an expensive product in limited supply because of the collection and processing steps from donated blood and because they cannot be frozen.8 In the United States, in 2018 alone, there were 2.4 million platelet doses (3 × 1011 to 6 × 1011 platelets each) administered at a cost of ∼$524 per dose. As they cannot be frozen, platelets have a useful life of 4 to 5 days and therefore not easily available in remote locations. Also, there is the possibility of bacterial or viral contamination of platelet products from blood collections.9 Thus, a safe, synthesized product that can serve as a substitute for platelets and that can be kept frozen is most desirable. Culture-derived platelets hold great potential for providing an abundant platelet supply,10,11 but many problems remain.12 In addition to platelet transfusions, there is a need to enhance platelet biogenesis in patients with thrombotic deficiency (such as is encountered during the treatment of cancer8 ) or excessive bleeding due to trauma.
We have previously shown that human MkMPs (huMkMPs; which can be stored frozen without loss of function13 ) specifically target human CD34+ HSPCs (huHSPCs) in vitro, leading to fate modification of huHSPCs toward Mk differentiation in the absence of TPO.4,13 Specifically, coculture of MkMPs with huHSPCs leads to the formation of mature Mks displaying characteristic PPT structures and synthesizing both α- and dense granules without exogenous TPO.4,13 Based on these findings, we hypothesized that huMkMPs maintain their in vitro biological activity in vivo and can thus serve as a potential therapeutic agent in transfusion medicine. To pursue this hypothesis, we first demonstrated that, in vitro, huMkMPs can target murine HSPCs (muHSPCs) to trigger murine Mk differentiation. Using a wild-type (WT) murine model, we further demonstrated that infusion of huMkMPs promotes de novo murine platelet biogenesis and can ameliorate antibody-induced murine thrombocytopenia.
Materials and methods
Several, previously detailed, experimental procedures can be found in the supplemental Materials and methods. They include chemicals and reagents, in vitro human Mk culture derived from mobilized peripheral blood (MPB) CD34+ HSPCs, and isolation of human huMkMPs.
Isolation of muHSPCs
All procedures involving mice were approved by the University of Delaware (UD) Institutional Animal Care and Use Committee and are in agreement with the Guide for the Care and Use of Laboratory Animals.14 All mice were housed with day and night stimulation at UD’s animal facility with access to food and water. Four- to 6-week-old female BALB/c mice (Jackson Laboratory) were euthanized via CO2 asphyxiation followed by cervical dislocation to ensure death. Mice were prepared for organ collection by spraying with 70% ethanol. Femurs were extracted and placed in RPMI 1640 (ThermoFisher) with 10% fetal bovine serum and 1% antibiotic-antimycotic (ThermoFisher) to prevent neutrophil activation. Femurs were decontaminated by triple washes with phosphate-buffered saline (PBS) containing 1% antibiotic-antimycotic. Once decontaminated, femur epiphyses were cut and flushed with RPMI 1640 using a 20-gauge needle attached to a 12-mL syringe. The effluent was collected and passed through a pre-separation filter (30 µm; Miltenyi). This process continued until the bone appeared white on the surface, indicating that the bone was empty of bone marrow.
Next, cells were collected at 300 g centrifugation for 10 minutes, followed by red blood cell (RBC) depletion with the treatment of ACK buffer (0.15 M NH4Cl, 10 mM KHCO3, and 0.1 mM Na2EDTA [pH 7.2–7.4]) for 5 minutes at 4°C.15 After that, cells were washed twice with PBS (containing 1% antibiotic-antimycotic). To isolate muHSPCs, lineage cells were depleted, using the Lineage Depletion Kit (Miltenyi), according to the manufacturer’s protocol.
Coculture of muHSPCs with huMkMPs
Coculture of huMkMPs and muHSPCs was performed in a 24-well Transwell plate to enhance the interaction between huMkMPs with muHSPCs. A Transwell membrane insert (No. 3470; Costar) was inserted into a well with 750 μL of growth medium (Iscove’s modified Dulbecco’s medium [IMDM]; 20% bovine serum albumin, insulin, transferrin; and 50 ng/μL of human stem cell factor [hSCF]) in the absence of TPO in the bottom compartment. muHSPCs (60 000) in 200 μL IMDM were seeded onto the membrane insert. After 20 minutes, huMkMPs were added to the membrane at a ratio of 30 huMkMPs per muHSPC. Both short-term (3-5 hours) and long-term (5 days) cocultures were performed.
For short-term cocultures, huMkMPs were prestained with the lipophilic dye PKH26 and cytosolic dye carboxyfluorescein diacetate succinimidyl ester (CFSE). After 3 or 5 hours of coculture, cells were harvested, and huMkMP uptake by muHSPCs was examined via confocal microscopy (LSM880; Zeiss).
For long-term cocultures, cells were cultured in Transwell plates16 for 24 hours, followed by transferring to the lower compartment of the plate. muHSPCs without addition of huMkMPs served as the vehicle control, whereas muHSPCs cultured in medium supplemented with 50 ng/mL of human TPO served as the positive control. At day 5, some cells were harvested for CD41 and ploidy flow-cytometric analysis15 ; some other cells were stained with anti-CD41 antibody for live-cell confocal microscopy. Some cells were fixed and stained for β-tubulin I (TUBB1), von Willebrand factor (VWF), and nucleus (4′,6-diamidino-2-phenylindole [DAPI]) imaging, as described.4 Images were acquired using a Zeiss LSM 880 multiphoton confocal microscope.
Chinese hamster ovary cell-derived empty vesicles
Chinese hamster ovary microvesicles (CHO-MVs) were generated from CHO membranes using the protocol of Fang et al,17 which yields uniform membrane vesicles with minimal loss to membrane functionality.18 In brief, CHO cells were collected from a standard CHO culture19 at day 2, stained with PKH26, and lysed with a Dounce homogenizer (Kimble) and hypotonic lysis buffer with protease inhibitor (p8340; Sigma). PKH26-stained cell membranes were isolated using differential ultracentrifugation. The final membrane pellet was resuspended in 700 µL filtered PBS and extruded through a 400-nm polycarbonate membrane (Avanti Lipid Extruder) at 55°C. The concentration of extruded CHO-MVs was quantified by flow cytometry, as for MkMPs.4
Murine experiments: in vivo impact of intravenously injected huMkMPs
We chose to use 4- to 6-week-old female BALB/c mice, because the variation of platelet levels of female mice is less than in male mice20 and would enable reaching statistically significant levels faster. To evaluate the biological effectiveness of huMkMPs, huMkMPs were injected into untreated mice or mice treated to induce thrombocytopenia. For untreated mice, 2 × 106 or 6 × 106 huMkMPs or 6 × 106 CHO-MVs in 100 μL saline or saline only were injected intravenously via the tail vein. CHO-MVs were used as an additional control. At 72 hours after huMkMP injection, 10 μL of blood was collected retro-orbitally, and platelet counts were measured by flow cytometry. Platelets were gated using the forward-scatter and side-scatter gates and standardized with microbeads (0.2-1.34 μm). The gating was confirmed using fluorescein isothiocyanate rat anti-mouse CD41(BD 553848) to distinguish between noise and platelet events. Control experiments confirmed that platelet counting is not affected by the presence of huMkMPs.
In another set of experiments, 6 × 106 PKH26-labeled huMkMPs, or saline control were administrated to BALB/c mice. Tissue, including bone marrow (BM), lung, liver, and spleen were collected and stored in 3-mL RPMI buffer with 10% FBS after 4 hours for single-cell isolation21 followed by RBC depletion. Harvested cells were stained with anti-CD41, -CD117, or -CD45 antibody and analyzed via flow cytometry.
To induce thrombocytopenia, LEAF rat anti-mouse CD41 antibody (MWReg30, Biolegend), at a dose of 1 μg/g murine body weight, was injected once intraperitoneally to induce thrombocytopenia.22 Eight hours after injection, mice (untreated or thrombocytopenic) received 2 × 106 huMkMPs in 100 μL saline or saline only via intravenous tail vein injection. Blood (10 μL) was collected retro-orbitally, and platelet counts were measured by flow cytometry 24 hours after antibody injection. Reticulated-platelet levels were measured as described,22 with a thiazole orange assay (BD Retic-Count).
Biodistribution experiments
PKH26-stained huMkMPs (106) or CHO-MVs in 100 μL PBS were injected intravenously into the tail vein of the mice, along with saline-only (PBS) controls. Four hours after injection, 1 mouse from the control group and 3 mice from the huMkMP-injected group were euthanized through CO2 asphyxiation, and the following tissues were surgically excised from each mouse: blood, BM of the femur, brain, heart, kidney, liver, lung, and spleen. The net weights of each mouse and all tissues, except blood, were recorded and the tissues were stored in 3 mL of PBS on ice. This procedure was repeated with mice collected 24 hours after injection of huMkMP, CHO-MV, or saline-only control. All tissues were processed within 24 hours of collection. Each tissue, except for blood, was lysed and homogenized with a coarse Dounce homogenizer in 1.5 mL of hypotonic lysis buffer (20 mM Tris-HCl, 10 mM KCl, 2 mM MgCl2, and 0.5% v/v p8340 protease inhibitor [pH 7.5]). Each tissue homogenate (200 μL) was added to a well of a black, opaque 96-well plate, and absolute fluorescence levels were measured with a SpectraMax i3x plate reader (excitation, 547 nm). Fluorescence levels of each tissue from treated mice were assessed as the difference between the absolute fluorescence of experimental mice and fluorescence from tissues of control mice (saline). These values were normalized by (1) overall tissue weight and (2) tissue weight as a fraction of the mouse weight. Normalization by tissue weight accounted for tissue-specific weight variation within each mouse. The mean fluorescence intensity (MFI) of each tissue was divided by the respective tissue masses (weight) to yield MFI per gram tissue mass.
Statistical analysis
Data are presented as the mean ± standard error of the mean (SEM). Unpaired Student t tests of all data were performed.
Results
In vitro, huMkMPs target muHSPCs and induce their Mk differentiation and expansion in the absence of TPO
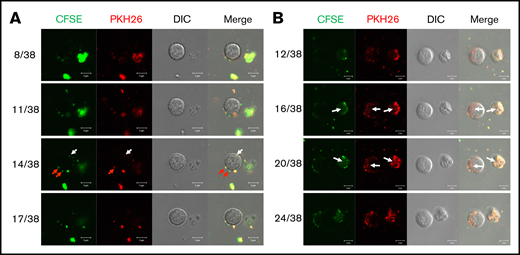
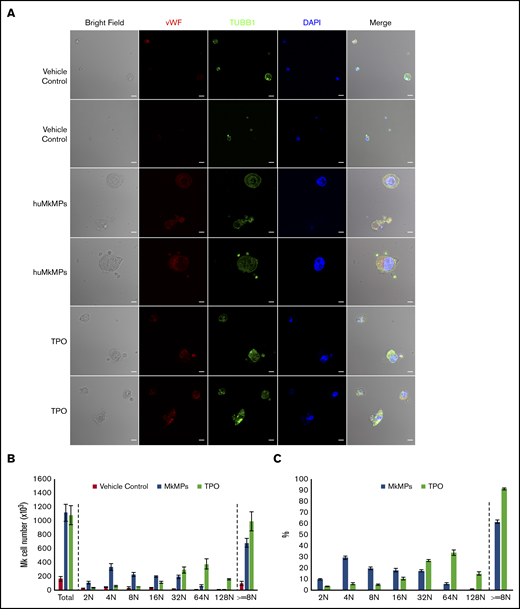
We have previously shown that, in vitro, huMkMPs induce and promote Mk differentiation of huHSPCs without stimulation by TPO, whereby the resulting Mks form functional PPTs that synthesize both α and dense granules.4 Later, we showed that huMkMPs target only huHSPCs and no other physiologically or ontologically related cells.13 In the present study, to test whether huMkMPs target and induce muHSPCs to differentiate into murine Mk cells, we first examined the uptake of huMkMPs by muHSPCs via short-term cocultures. We generated huMkMPs from day-12 primary Mk cultures started with frozen, human MPB CD34+ cells, as described.4,13 huMkMPs were stained with CFSE, a green cytosolic dye staining surface and cytosolic proteins by covalently binding to amines, and PKH26, a lipophilic, membrane-staining dye, and were cocultured with muHSPCs. We acquired confocal microscopy images at multiple z-planes, aiming to determine whether huMkMPs recognize, attach to, and are possibly internalized by muHSPCs. We examined 70 cells at different time points to capture different stages of interactions between huMkMPs and muHSPCs. huMkMPs recognized, targeted, and attached to muHSPCs after 3 hours of coculture (Figure 1A) and were taken up by muHSPCs within 5 hours (Figure 1B). At 3 hours, at all z-planes, several stained huMkMPs appeared attached to muHSPCs (red arrows), and some were possibly internalized and localized just inside (white arrows) the cellular membrane of muHSPCs. By hour 5, most MkMPs were localized inside the murine cells, as indicated by the different fluorescent patterns from the stained huMkMPs at different z-planes at the same location of murine cells. Several of these stained huMkMPs released their content (stained green) into the murine cells, as indicated by comparison of the green fluorescent regions within the murine cells at different z-planes (eg, plane 16 of 38 vs 20 of 38). These patterns of release of huMkMP green-stained content into target cells resemble the previously reported patterns of huMkMP content released into the huHSPCs.13 We concluded that huMkMPs recognize and are internalized by muHSPCs in a manner apparently similar to that by which they recognize and are internalized by huHSPCs. Next, to examine whether huMkMPs are capable of triggering muHSPC Mk differentiation as they do for huHSPCs, we cocultured huMkMP and muHSPCs (without TPO) for 5 days. Our previous data have shown that, based on our culture protocol, murine Mk cells reach the highest polyploidy state after 4 to 6 days of in vitro culture with TPO.15,22 In this study, day 5 cells were fixed and stained for TUBB1, VWF, and nucleus (DAPI). As shown in Figure 2A and supplemental Figure 1, we identified large Mk cells with strong VWF expression and proplatelet and platelet formation, both from huMkMP coculture and TPO culture. Furthermore, strong CD41 expression was detected on cells using live-cell imaging of either huMkMPs coculture or TPO culture, but not of vehicle control (supplemental Figure 2). Next, we examined muMk maturation via ploidy analysis. The number of total and polyploid (≥8N) Mk cells from the coculture with huMkMPs increased significantly (by 6.8- to 7.0-fold) compared with the vehicle control cells. Notably, whereas a similar number of total Mks were produced at day 5 in huMkMPs with TPO culture (Figure 2B), muMks from the huMkMP-induced culture exhibited less mature characteristics (4-32N) than those from the TPO-induced culture (16-128N; Figure 2C). Representative plots of ploidy analysis via flow cytometry are shown in supplemental Figure 3. Previously, we have shown that huMkMPs were able to induce Mk differentiation and promote cell proliferation of huHSPCs (4 (Fig5B)). Similarly, our data in the current study demonstrate that huMkMPs target and trigger Mk differentiation and proliferation of muHSPCs and proplatelet and platelet production.
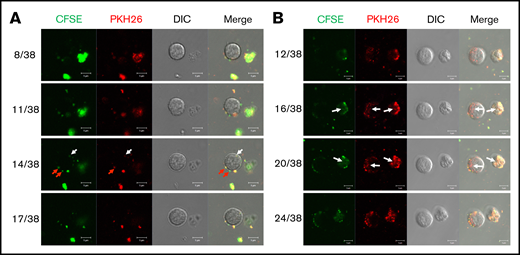
Coculture of huMkMPs and muHSPCs in vitro. muHSPCs were cocultured for up to 5 hours with huMkMPs stained with CFSE and PKH26. The cells were examined by confocal microscopy at 3 (A) and 5 (B) hours after the start of coculture, and fluorescence images were taken at different confocal planes. The numbers represent the specific slice (Z-stack) number (left)/total number of slices (right). Red arrows indicate huMkMPs binding to muHSPCs; white arrows indicate huMkMPs that have been internalized by muHSPCs.
Coculture of huMkMPs and muHSPCs in vitro. muHSPCs were cocultured for up to 5 hours with huMkMPs stained with CFSE and PKH26. The cells were examined by confocal microscopy at 3 (A) and 5 (B) hours after the start of coculture, and fluorescence images were taken at different confocal planes. The numbers represent the specific slice (Z-stack) number (left)/total number of slices (right). Red arrows indicate huMkMPs binding to muHSPCs; white arrows indicate huMkMPs that have been internalized by muHSPCs.
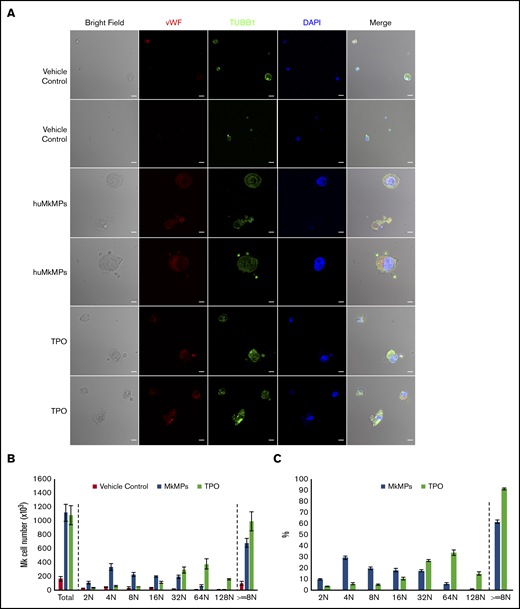
Coculture of huMkMPs and muHSPCs promotes murine megakaryopoiesis. Sixty thousand muHSPCs were cocultured for 5 days with huMkMPs only, with 50 ng/mL TPO, or without any supplements (vehicle control). Cells were harvested and fixed at day 5 and (A) stained for expression of VWF, TUBB1, and nucleus (DAPI), via confocal microscopy along with bright-field imaging. Some cells were analyzed by flow cytometry to count Mk cells (B) or to measure the percentage of Mk cells (C) in the various ploidy classes. The bar represents 10 μm; error bars represent the SEM (n = 4).
Coculture of huMkMPs and muHSPCs promotes murine megakaryopoiesis. Sixty thousand muHSPCs were cocultured for 5 days with huMkMPs only, with 50 ng/mL TPO, or without any supplements (vehicle control). Cells were harvested and fixed at day 5 and (A) stained for expression of VWF, TUBB1, and nucleus (DAPI), via confocal microscopy along with bright-field imaging. Some cells were analyzed by flow cytometry to count Mk cells (B) or to measure the percentage of Mk cells (C) in the various ploidy classes. The bar represents 10 μm; error bars represent the SEM (n = 4).
Infusion of huMkMPs enhance murine platelet levels in both normal and thrombocytopenic mice
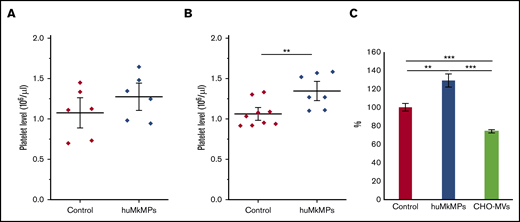
We hypothesized that the in vitro biological activity of MkMPs, that is, their ability to target and program muHSPCs toward Mk differentiation is retained in vivo. To test this hypothesis, we first examined the biological effect of intravenous administration of huMkMPs in healthy WT BALB/c mice. In brief, 2 different doses of huMkMPs (2 × 106 or 6 × 106) were injected intravenously into the mice via the tail vein. At 72 hours after huMkMP injection, blood was collected to count the murine platelet concentrations. We chose 72 hours as the blood collection time point based on the assumption that 3 days would be a reasonable period to see the impact of de novo platelet biogenesis from muHSPCs. Upon administration of 2 × 106 huMkMPs, murine platelet levels were elevated by 18.5%, but not in a statistically significant way (Figure 3A), but injection of 6 × 106 huMkMPs significantly elevated murine platelet levels by 26.7% (Figure 3B). These data (Figure 3) suggest that huMkMPs target muHSPCs to induce murine Mk differentiation and platelet biogenesis. As an additional control, to exclude the possibility that the effect of huMkMPs on murine platelet counts possibly derives from a secondary reactive thrombocytosis related to the injection of cell membranes, 6 × 106 CHO cell–derived empty MVs were injected into the BALB/c mice. As shown in Figure 3C, CHO-MVs did not increase (in fact, they decreased) platelet levels at 24 hours after injection.
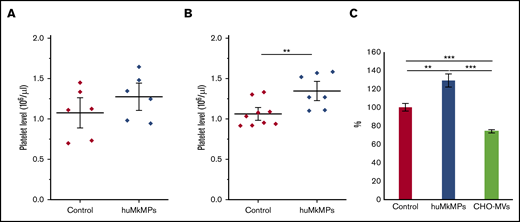
Intravenous administration of huMkMPs increases in vivo platelet concentrations in WT mice 72 hours after administration. huMkMPs, 2 × 106 (A) or 6 × 106 (B), were administered intravenously into WT BALB/c mice via the tail vein. Murine platelet levels of control mice (untreated) or mice treated with MkMPs were measured 72 hours after huMkMP injection. (C) huMkMPs (6 × 106; n = 8), CHO-MVs (6 × 106; n = 2), or saline control (n = 10) were administered intravenously to WT mice via the tail vein, and murine platelet numbers were measured as percentage of the control at 24 hours. Error bars represent the SEM. **P < .01; ***P < .001.
Intravenous administration of huMkMPs increases in vivo platelet concentrations in WT mice 72 hours after administration. huMkMPs, 2 × 106 (A) or 6 × 106 (B), were administered intravenously into WT BALB/c mice via the tail vein. Murine platelet levels of control mice (untreated) or mice treated with MkMPs were measured 72 hours after huMkMP injection. (C) huMkMPs (6 × 106; n = 8), CHO-MVs (6 × 106; n = 2), or saline control (n = 10) were administered intravenously to WT mice via the tail vein, and murine platelet numbers were measured as percentage of the control at 24 hours. Error bars represent the SEM. **P < .01; ***P < .001.
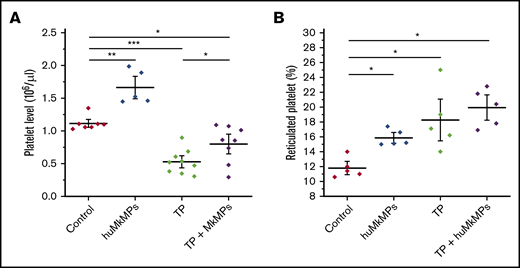
Next, we investigated the potential of huMkMPs for treating murine thrombocytopenia. Based on our previous study,22 an anti-CD41 antibody (1 µg/g of body weight) was intraperitoneally administrated to mice to induce thrombocytopenia. Expectedly, platelet levels dropped dramatically within 24 hours and did not fully recover until day 5.22 We then chose to examine whether huMkMPs would ameliorate the drop in platelet concentrations by intravenous administration via the tail vein of 2 × 106 huMkMPs 8 hours after thrombocytopenia induction. We chose the lower of the 2 doses examined in the experiments of Figure 3 for practical reasons: namely, the ability to cost and time effectively generate a large enough number of huMkMPs to treat a large cohort of mice. At 24 hours after thrombocytopenia induction (the nadir of platelet concentrations with this induced-thrombocytopenia protocol22 ), as shown in Figure 4A, platelet levels dropped 47% in thrombocytopenic mice. Administration of 2 × 106 huMkMPs to untreated mice significantly increased murine platelet levels (by 49%) 16 hours after huMkMP administration, whereas murine platelet levels in thrombocytopenic mice were significantly elevated (by 51%), although the levels were not restored to the those in the untreated mice. It was somewhat unexpected that administration of the lower dose of 2 × 106 huMkMPs into untreated mice would increase murine platelet levels by 49% after 16 hours, given the results shown in Figure 3A. This comparison then would suggest that huMkMPs induce murine platelet biogenesis very quickly and that, by 72 hours, the platelet levels appeared to be dropping toward homeostatic levels.
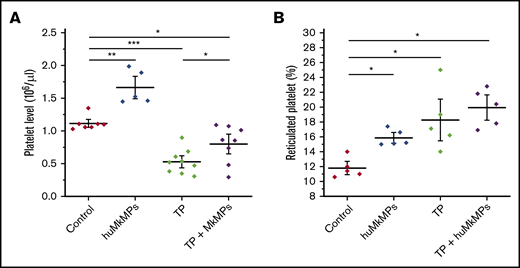
Intravenous administration of huMkMPs into untreated WT mice increases murine platelet concentrations and ameliorates thrombocytopenia (TP) in thrombocytopenic mice 16 hours after administration. TP was induced by intraperitoneal administration of anti-CD41 antibody. After 8 hours, 2 × 106 huMkMPs were administered intravenously into untreated or thrombocytopenic mice via the tail vein. (A) Murine platelet levels were measured 24 hours after antibody injection (16 hours after huMkMP administration) of untreated mice (n = 7), mice treated with huMkMPs (n = 5), thrombocytopenic mice (n = 9), or thrombocytopenic mice treated with MkMPs (n = 8). (B) The number of reticulated (newly synthesized) platelets was measured 24 hours after antibody injection (16 hours after huMkMP administration) by flow cytometry for a subset of mice in the 4 murine cohorts in panel A. The data are presented as the percentage of the total platelet count. Error bars represent the SEM. *P < .05; **P < .01; ***P < .001.
Intravenous administration of huMkMPs into untreated WT mice increases murine platelet concentrations and ameliorates thrombocytopenia (TP) in thrombocytopenic mice 16 hours after administration. TP was induced by intraperitoneal administration of anti-CD41 antibody. After 8 hours, 2 × 106 huMkMPs were administered intravenously into untreated or thrombocytopenic mice via the tail vein. (A) Murine platelet levels were measured 24 hours after antibody injection (16 hours after huMkMP administration) of untreated mice (n = 7), mice treated with huMkMPs (n = 5), thrombocytopenic mice (n = 9), or thrombocytopenic mice treated with MkMPs (n = 8). (B) The number of reticulated (newly synthesized) platelets was measured 24 hours after antibody injection (16 hours after huMkMP administration) by flow cytometry for a subset of mice in the 4 murine cohorts in panel A. The data are presented as the percentage of the total platelet count. Error bars represent the SEM. *P < .05; **P < .01; ***P < .001.
To test the hypothesis that huMkMPs induce de novo biogenesis of murine platelets (rather than increasing their stability and half-life in circulation), we examined whether injection of 2 × 106 huMkMPs into thrombocytopenic or untreated mice would increase the percentage of newly synthesized (reticulated) platelets. As shown in Figure 4B, administration of 2 × 106 MkMPs to untreated mice significantly enhanced the percentage of reticulated platelets from the steady-state level of 11.8% to 15.9%. Although the induction of thrombocytopenia increased the level of reticulated platelets to 18.3%, administration of huMkMPs increased the reticulated platelet levels to 19.9%. The data in Figure 4B support the hypothesis that the observed increases in platelet concentrations are a result of the interaction between huMkMPs and muHSPCs, giving rise to newly synthesized murine platelets.
Biodistribution of huMkMPs in the WT murine model
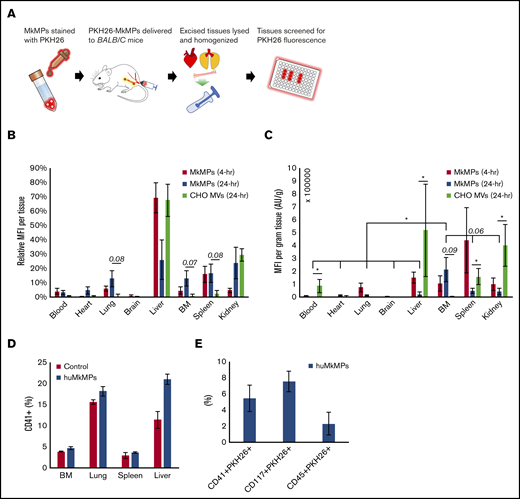
To complement the data reported above, we investigated the fate of huMkMPs transfused into mice. In brief, BALB/c mice were intravenously administered with 6 × 106 PKH26-stained huMkMPs or 6 × 106 PKH26-stained CHO-MVs in filtered PBS via the tail vein. As the properties of the CHO-MV membranes are very distinct from those of huMkMPs, CHO-MVs were again used as a control for these biodistribution studies. Another control group of mice were given an equivalent volume of PBS only. To examine the in vivo biodistribution of the PKH26-huMkMPs or PKH26-CHO-MVs, 2 sets of mice injected with huMkMPs (for collection of tissues at 4 and 24 hours) and 1 set of mice infused with CHO-MVs (for tissue collection at 24 hours) were euthanized, and the PKH26 signals from various tissues (liver, BM, femurs, blood, spleen, kidney, brain, lungs, and heart) were examined (Figure 5A); the relative MFI of PKH26 for each tissue is shown in Figure 5B. PKH26-MFI was greatest in the liver and spleen 4 hours after administration of PKH26-stained huMkMPs. As both of these tissues directly interact with circulating blood, temporary localization of PKH26-stained huMkMPs was expected in these tissues, and thus, this result was expected based on historical biodistribution data of liposomes administered to BALB/c mice.23,24 These findings also mimic the results of other biodistribution studies involving liposomes and nanoparticles, where the liver, kidney, and spleen exhibited the greatest initial uptake of labeled particles.25-28 After 24 hours, the relative MFI was reduced in the liver but increased (relative to t = 4 hours) in the BM, kidney, lung, and heart by two- to fivefold (Figure 5B), indicating possible target specificity of the huMkMPs. Unlike huMkMPs, CHO-MVs exhibited a different pattern at 24 hours (Figure 5B), with ∼70% of stained CHO-MVs in liver and ∼30% in kidney. The biodistribution of the CHO-MVs largely mimicked the patterns exhibited by other membrane vesicles and liposomes in published biodistribution studies.25-28 These results also suggest that the surface moieties on MkMPs vs those of CHO-MVs could possibly play a role in how these vesicles interact with different cells and tissues.
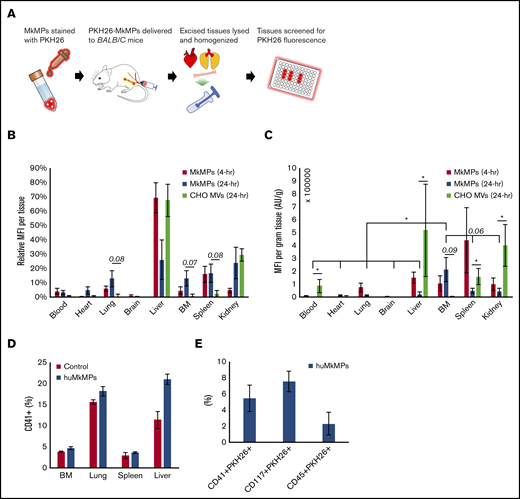
In vivo biodistribution of PKH26-labeled huMkMPs administered to untreated WT mice and huMkMP colocalization with murine blood cells. Murine tissues were excised, homogenized, and analyzed for fluorescence (SpectraMax i3x) 4 and 24 hours after administration of huMkMPs and CHO-MVs. (A) Experimental schema for measuring tissue-specific fluorescence. (B) MFI in each excised tissue relative to total fluorescence in all tissues (n = 6 mice per MkMP group; n = 3 mice in CHO-MV group). (C) MFI of each tissue normalized by tissue weight is shown. (D) PKH26-labeled huMkMPs (6 × 106), or saline control were administered to BALB/c mice. Tissues, including BM, lung, liver, and spleen, were collected after 4 hours for single-cell isolation. Harvested cells were stained with anti-CD41 antibody and analyzed via flow cytometry (n = 2). (E) Single cells isolated from the liver were analyzed by flow cytometry for colocalization of PKH26 signal with CD41, CD117, or CD45 signals (n = 2). Error bars represent the SEM. The unpaired 2-tailed Student t test was used to determine statistical significance. *P < .05. Higher P values of some comparisons are displayed over the bars.
In vivo biodistribution of PKH26-labeled huMkMPs administered to untreated WT mice and huMkMP colocalization with murine blood cells. Murine tissues were excised, homogenized, and analyzed for fluorescence (SpectraMax i3x) 4 and 24 hours after administration of huMkMPs and CHO-MVs. (A) Experimental schema for measuring tissue-specific fluorescence. (B) MFI in each excised tissue relative to total fluorescence in all tissues (n = 6 mice per MkMP group; n = 3 mice in CHO-MV group). (C) MFI of each tissue normalized by tissue weight is shown. (D) PKH26-labeled huMkMPs (6 × 106), or saline control were administered to BALB/c mice. Tissues, including BM, lung, liver, and spleen, were collected after 4 hours for single-cell isolation. Harvested cells were stained with anti-CD41 antibody and analyzed via flow cytometry (n = 2). (E) Single cells isolated from the liver were analyzed by flow cytometry for colocalization of PKH26 signal with CD41, CD117, or CD45 signals (n = 2). Error bars represent the SEM. The unpaired 2-tailed Student t test was used to determine statistical significance. *P < .05. Higher P values of some comparisons are displayed over the bars.
To account for different tissue masses, the MFI of each tissue was normalized by the excised tissue weight. The tissue weight-normalized MFI from the huMkMP-infused group was significantly greater in the BM than in any other excised tissue 24 hours after huMkMP administration, except perhaps for spleen and kidney, which were within the margin of significance (P = .06; Figure 5C). Among all tissues, only the ΒΜ demonstrated a large increase in normalized MFI; the heart exhibited a slight increase (Figure 5C). Similar to Figure 5B, higher normalized MFI from CHO-MVs was shown in liver, kidney, spleen, and circulating blood (Figure 5C). Note that storage of tissues in 3 mL of PBS for a few hours before processing would to some extent deplete the tissues of nonadherent blood cells.
Based on the different pattern of biodistribution exhibited by huMkMPs and CHO-MVs (Figure 5B) and the fast response of de novo platelet biogenesis (Figure 4), we hypothesized that huMkMPs can target muHSPCs or more differentiated cells in vivo. To examine this hypothesis, 6 × 106 PKH26-labeled huMkMPs, or saline control were administrated to BALB/c mice. BM, lung, liver, and spleen were chosen for examination based on the higher relative PKH26-MFI at 24 hours (Figure 5B). Tissues were collected after 4 hours, stored in 3-mL RPMI buffer with 10% FBS, and then processed for single-cell isolation and RBC depletion. Harvested single cells were stained with anti-CD41, -CD117, or -CD45 antibody and analyzed via flow cytometry. Somewhat unexpectedly, in addition to BM, the liver, lung, and spleen appeared to be a reservoir of a good number of CD41+ (Mk) cells (Figure 5D), despite the storage (that would remove loosely adherent blood cells) of these tissues in 3-mL RPMI buffer before processing. For example, almost 20% of the single cells from the processing of the liver tissue from the mice infused with huMkMPs were CD41+. It could be assumed that these were circulating CD41+ progenitors and immature Mk cells trapped or retained in the liver and similarly in the other tissues. Although not statistically significant because of the small number of mice used (n = 2), upon huMkMP infusion, the percentage of CD41+ cells generally increased in each tissue, especially in the liver and lung (Figure 5D). Furthermore, we examined the colocalization of signal from the PKH26+ huMkMPs and murine HSPCs (CD117/c-KIT+), CD41+ cells, and hematopoietic (CD45+) cells in the liver, which had the highest fraction of CD41+ cells, thus making data outcomes and their analysis easier and more robust. As shown in the Figure 5E, a significant fraction of CD41+ cells were CD41+PKH26+. This fraction is one-fourth (25%) the CD41+ cells in the liver, which was calculated as follows: 5% of single cells from the liver processing were CD41+PKH26, whereas 20% of single cells from the liver processing were CD41+ cells (Figure 5D). Similarly, there were significant numbers of cells that were CD117+PKH26+ (∼8% of single cells from the liver processing; 45% of the CD117+ cells) and CD45+PKH26+ (2%; 6% of the CD45+ cells). These data indicate that CD117+, CD41+, and generally immature hematopoietic (CD45+) cells were targeted by huMkMPs. We were not able to identify colocalization of PKH26 signal with CD41+ cells in the BM, possibly due to the low frequency of CD41+ cells in the BM (Figure 5D) and the relatively low number of PKH26+ huMkMPs in the BM (Figure 5B-C), thus rendering such colocalization events below the flow-cytometric detection limits and similarly for colocalization of PKH26 signal with CD117+ or CD45+ cells in the BM. Nevertheless, these results and notably the data in Figure 5D-E suggest that huMkMPs target CD117+, CD41+, and CD45+ cells (collectively, progenitor cells), and, together with the data of Figures 5B-C, that the targeting is localized in the liver, lung, BM, and spleen. Liver and lung emerge, then, as significant sites of CD41+-cell biogenesis upon PKH26+ huMkMPs infusion.
Discussion
We demonstrated that, in vitro, huMkMPs can target muHSPCs, the same as they target huHSPCs (Figure 1). This targeting induced Mk differentiation of muHSPCs and was comparable to TPO-induced differentiation (Figure 2). We have previously shown that huMkMPs recognize and target huHSPCs via huHSPC receptors that include CD11b, CD18, CD54, and CD43.13 Based on Basic Local Alignment Search Tool analysis, CD11b, CD18, CD54, and CD43 show protein-level conservation between human and murine cells of 86%, 90%, 67%, and 59%, respectively. It is possible that huMkMPs target muHSPCs by using the same receptors. We have also identified 2 mechanisms of how huHSPCs internalize huMkMPs: membrane fusion and endocytosis.13 Similar mechanisms may be engaged for the interactions between huMkMPs and muHSPCs.
Based on our in vitro studies, we then demonstrated that intravenous administration of huMkMPs into healthy or thrombocytopenic mice resulted in enhanced total platelet levels with newly synthesized platelets at 16 hours after huMkMPs injection (Figure 4B). It was somewhat surprising that huMkMP induced de novo murine platelet biogenesis so quickly, and this suggests that huMkMPs not only target muHSPCs but also more differentiated cells, to induce Mk differentiation. This hypothesis is supported by the current finding shown in Figure 5D-E together with our published finding that huMkMPs target and can induce Mk differentiation and enhanced survival of day-1 and -3 cultured human CD34+ cells.4 We also demonstrated that, in vivo, huMkMPs localize quickly (4 hours) and are highly enriched in the BM, lung, spleen, and kidney 24 hours after injection (Figure 5B-C). It has been reported that lungs and spleen are important reservoirs of Mk progenitors producing proplatelets and platelets.29 Our data suggest that huMkMPs retained in the spleen and lung (Figure 5B-C) target hematopoietic or Mk progenitors (Figure 5E) to rapidly induce de novo platelet biogenesis. It has been shown that HSPCs are also found in the adult liver,30 and although Mk cells could not be detected in the adult liver using 2-photon intravital microscopy,29 given the presence of HSPCs and the production of TPO in liver, Mk cells would be logically expected in the adult liver. Our data (Figure 5D-E) seem to confirm the presence of muCD41+ cells in the adult murine liver, where these and muHSPCs may be targeted by huMkMPs to produce platelets in the liver, as well.
The biological impact of huMkMPs on muHSPCs may be mediated by the delivery of huMkMP RNA, as is the case with huMkMPs targeting huHSPCs.13 We have recently reported that 2 of the most abundant microRNAs (miRs) in huMkMPs, miR-486-5p and miR-22-3p, are likely important in triggering Mk differentiation in huHSPCs.16 These 2 miRs are identically conserved on the murine genome,31 and their role appears to be conserved as well.32
Several methods have been explored for delivering therapeutic cargo to specific cells or tissues in vivo, but tissue- or cell-specific delivery remains a challenge, particularly for HSPCs.33 Viral vectors have high transfection efficiencies but could be toxic because of insertional mutagenesis and could elicit an immune response from the host.33 Other carriers, such as liposomes, have demonstrated effective delivery of cargo to different tissues in vivo, but target specificity is attained only by engineering the liposome’s surface.28,34 The use of EVs addresses some of the shortcomings of liposomes and viral vectors. Wen et al35 have demonstrated the innate targeting ability of intravenously administered 4T1 and 67NR exosomes to target breast-tumor cells in vivo. Tian et al28 demonstrated specific targeting of cerebral tissue with exosomes. Similar to our findings, prior studies have reported initial localization of labeled EVs in liver, kidneys, and spleen, with improved tissue-specific targeting occurring after a 24-hour circulation.28,35 However, Tian et al28 observed significant retention in the liver with extended circulation, with a several-fold increase in liver localization compared with the targeted tissue. In the present study, huMkMP localization in the BM was significantly or near significantly higher than all other tissues examined.
In vivo, Mks can produce between 2 000 and 11 000 platelets, but in vitro only a few Mk cells can produce platelets (typically, 10-100 per Mk).38-40 With current optimized practices, it is possible to produce 40 to 50 Mks per input HSPC (CD34+ cell).41 Thus, a single platelet transfusion unit will require almost 108 CD34+ cells, which is 50-fold higher than the number contained in a single umbilical cord blood (UCB) collection (2 × 106), or 50% of a single apheresis of MPB, typically 2 × 108 CD34+ cells. The availability and cost of such a large number of CD34+ cells and the small number of platelets or PLPs generated per CD34+ cell limit the scalability and the economic feasibility of platelet synthesizing. However, by focusing on manufacturing MkMPs from cultured Mks, the numbers look promising, and the process appears to be both feasible and cost-effective. Table 2 shows calculations of MkMP yields per input Mk or CD34+ cell collected from UCB or MPB, based on current performance metrics, including the use of biomechanical forces (shear) to enhance MkMP biogenesis by 47-fold compared with static MkMP production.4 These calculations suggest that the production of therapeutic doses of MkMPs is practical. With process optimization and the development of large-scale EV manufacturing methods,42 MkMP yields per input CD34+ cell would be expected to improve by at least 50- to 100-fold.
Based on these proof-of-concept findings and engineering analyses, huMkMPs appear to hold good potential to serve as a platelet substitute to ameliorate thrombocytopenias and to enhance platelet biogenesis in patients with thrombotic deficiency. In view of the fact that huMkMPs can be stored frozen,13 such an MkMP product could also resolve the issue of availability on demand at any location worldwide. Furthermore, based on our recent data16 showing that, in vitro, MkMPs can be used to effectively deliver exogenously loaded plasmid DNA and small RNAs to huHSPCs, our data reported herein, demonstrating in vivo effectiveness of huMkMPs in targeting HSPCs, would open up the possibility of using modified MkMPs for gene therapy applications to address hematologic abnormalities.
Original data may be obtained by e-mail to the corresponding author.
Acknowledgments
The authors thank Jeff Caplan and members of the Bioimaging Center (University of Delaware) for assistance with the confocal microscopy.
This project was supported by US National Science Foundation grant CBET-1804741.
Authorship
Contribution: E.T.P., C.E., C.-Y.K., and S.D. designed the study and analyzed the data; C.E., C.-Y.K., and S.D. carried out the experiments; and E.T.P., C.-Y.K., and S.D. wrote the manuscript.
Conflict-of-interest disclosure: E.T.P and C.-Y.K. are listed as inventors on a pending US/PCT patent application (publication US20170058262A1, application no. 15/308 221, PCT no. PCT/US15/31 388) on MkMP-based technologies. The rights to the pending patent belong to the 3 inventors of the patent. The remaining authors declare no competing financial interests.
Correspondence: Eleftherios T. Papoutsakis, Delaware Biotechnology Institute, 15 Innovation Way, Newark, DE 19711; e-mail: epaps@udel.edu.