

Key Points
Delayed CD4+ T-cell recovery leads to a risk for opportunistic infections for ≥1 year after CD19-directed CAR T-cell therapy.
Antimicrobial prophylaxis, immunoglobulin replacement, and growth factor use may minimize nonrelapse morbidity and mortality after axi-cel.

Abstract
Chimeric antigen receptor (CAR) T-cell therapy targeting CD19 has significantly improved outcomes in the treatment of refractory or relapsed large B-cell lymphoma (LBCL). We evaluated the long-term course of hematologic recovery, immune reconstitution, and infectious complications in 41 patients with LBCL treated with axicabtagene ciloleucel (axi-cel) at a single center. Grade 3+ cytopenias occurred in 97.6% of patients within the first 28 days postinfusion, with most resolved by 6 months. Overall, 63.4% of patients received a red blood cell transfusion, 34.1% of patients received a platelet transfusion, 36.6% of patients received IV immunoglobulin, and 51.2% of patients received growth factor (granulocyte colony-stimulating factor) injections beyond the first 28 days postinfusion. Only 40% of patients had recovered detectable CD19+ B cells by 1 year, and 50% of patients had a CD4+ T-cell count <200 cells per μL by 18 months postinfusion. Patients with durable responses to axi-cel had significantly longer durations of B-cell aplasia, and this duration correlated strongly with the recovery of CD4+ T-cell counts. There were significantly more infections within the first 28 days compared with any other period of follow-up, with the majority being mild-moderate in severity. Receipt of corticosteroids was the only factor that predicted risk of infection in a multivariate analysis (hazard ratio, 3.69; 95% confidence interval, 1.18-16.5). Opportunistic infections due to Pneumocystis jirovecii and varicella-zoster virus occurred up to 18 months postinfusion in patients who prematurely discontinued prophylaxis. These results support the use of comprehensive supportive care, including long-term monitoring and antimicrobial prophylaxis, beyond 12 months after axi-cel treatment.
Introduction
Chimeric antigen receptor (CAR) T-cell therapy targeting CD19 (CAR19) with axicabtagene ciloleucel (axi-cel) has significantly improved outcomes in the treatment of relapsed or refractory (R/R) large B-cell lymphoma (LBCL), including diffuse LBCL, primary mediastinal B-cell lymphoma, and transformed follicular lymphoma. Long-term follow-up of the patients treated with axi-cel as part of the pivotal ZUMA-1 trial demonstrated a durable objective response rate of 39% and OS of 47% after 3 years of follow-up.1,2 The acute side effects of CAR T-cell therapy, including cytokine release syndrome (CRS) and neurotoxicity (immune effector cell–associated neurotoxicity syndrome [ICANS]), have been well described, resulting in consensus guidelines on their management.3 However, the infectious risks and delayed effects of CAR T-cell therapies, including axi-cel, have been less comprehensively reported, despite increasing use.
Severe cytopenias (grade ≥3) are common in the first 28 days following the administration of lymphodepletion chemotherapy and subsequent axi-cel infusion.4 However, in ZUMA-1 and subsequent institutional cohorts, up to 17% of patients had a persistent severe cytopenia at 3 months postinfusion (12.5% neutropenia, 7% thrombocytopenia, 3.6% anemia) without evidence of bone marrow dysplasia or disease relapse.1,5-7 In addition to myelosuppression, B-cell aplasia, presumably due to on-target CAR19 effects, led to hypogammaglobulinemia and IV immunoglobulin (IVIG) use in up to 45% of patients.5,8
In institutional cohorts of patients treated with CAR19, up to 45% developed infections within the first 90 days post–CAR T-cell infusion.6,8,9 A majority of these infections occurred within the first 28 days and were classified as nonsevere (grade <3 or not requiring hospitalization). These same studies identified severe CRS and ICANS, as well as the use of tocilizumab and corticosteroids, as risk factors for infection within the first 28 days.
Here, we report on the timing and incidence of hematologic recovery, immune reconstitution, and infectious complications in the immediate and long-term follow-up of patients treated with axi-cel for R/R LBCL. In conjunction with the reported literature, we provide data-driven guidance for antimicrobial prophylaxis and supportive care measures as part of ongoing care for patients post–axi-cel.
Methods
Patients and data collection
Adult patients with R/R CD19+ LBCL treated with axi-cel at Stanford University’s Cancer Institute between 1 September 2017 and 1 March 2019 were included in the current analysis. Each patient consented to an Institutional Review Board–approved database and biorepository protocol prior to initiating therapy. Patients’ electronic medical records were reviewed to abstract information on patient and disease characteristics, laboratory data, infectious complications, CAR19-associated toxicities, and clinical events. B- and T-lymphocyte subsets were quantified from fresh peripheral blood samples using a validated research flow cytometry panel containing an anti-FMC63 antibody to identify CAR+ T cells at all follow-up visits. Antigen-specific immunoglobulin G (IgG) antibody titers were assessed using frozen plasma samples and direct enzyme-linked immunosorbent assay (supplemental Methods).
Lymphodepletion chemotherapy and adoptive transfer of CD19-directed CAR T cells
Patients received 1 cycle of lymphodepletion (LD) chemotherapy consisting of fludarabine, 30 mg/m2 per day, and cyclophosphamide, 500 mg/m2 per day, on days −5, −4, and −3, followed by axi-cel infusion with a CAR+ cell dose of 2 × 106 cells per kilogram of body weight on day 0, as in ZUMA-1.4
Supportive care and monitoring
Adverse events were graded per the Common Terminology Criteria for Adverse Events (CTCAE) v5.0. The severity of CRS and neurotoxicity was graded based on retrospective review per American Society for Transplantation and Cellular Therapy consensus criteria.3 Treatment with tocilizumab (8 mg/kg IV per dose) and/or corticosteroids (dexamethasone, 10 mg IV per dose or equivalent) was used for any patient who developed grade ≥2 CRS and/or neurotoxicity, respectively. These guidelines were consistent with the CARTOX working group recommendations and led to earlier intervention on average than was used for the ZUMA-1 phase 2 clinical trial.10 Institutional standard practices for monitoring and supportive care are outlined in supplemental Methods.
Infection categorization and grading
Infectious events were recorded using previously established Blood and Marrow Transplant Clinical Trials Network criteria.11,12 Events were classified using a microbiologic or histopathologic test or clinicoradiographic syndrome, and day of onset was defined as the day on which the diagnostic test was performed. Bacterial infections were categorized as a bacteremia or a site-specific infection. Repeated positive cultures for the same organism were considered a second event if they occurred >21 days after the initial event and intervening cultures were negative. Site-specific infections were defined by a positive culture of a normally sterile site or by culture and clinical or radiographic evidence of tissue invasion of a nonsterile site. Fungal infections were classified as proven or probable (eg, pulmonary nodules) based on the 2008 revised criteria for invasive fungal disease.13 Fever of undetermined origin (whether neutropenic or not) within the first 28 days was excluded.
Infection severity was graded according to CTCAE v5.0 specific to the event of interest. Severe infections (grade ≥3) were those that required IV antimicrobial therapy and/or hospitalization or were associated with life-threatening symptoms or invasive interventions, in keeping with previously reported categorizations.6,8,9
Statistical analysis
Fisher’s exact test and Pearson’s χ2 test were used to assess associations between categorical variables. Blood counts and lymphocyte subset dynamics over time were compared using the Kruskal-Wallis test; each time point was treated as an independent group. Median progression-free survival and overall survival (OS) for all patients in the study were estimated by the Kaplan-Meier (K-M) method, and 2-sample comparisons were made using the log-rank test. Nonrelapse mortality (NRM) was estimated by cumulative incidence time-to-event method.
All clinical and laboratory data were censored at the date of last follow-up. The durations of grade ≥3 cytopenias, B-cell aplasia, and cumulative infection incidence were modeled as event-free survival estimates using the K-M method, where disease progression or relapse and death were defined as competing events. Count recovery was defined as the first of 3 consecutive days with a given count value above the relevant cut point. To evaluate factors associated with the durations of severe cytopenias, clinical responses to axi-cel and age (split by cohort median) were treated as dichotomous categorical variables to perform univariate and stepwise multivariate Cox proportional-hazards regression and compared using the likelihood ratio test.
Infection densities were calculated as the mean number of events per 100 patient-days at risk within a set period of time postinfusion. To compare infection densities between time periods and groups, the incidence rate ratio (IRR) and 95% confidence interval (CI) were calculated using the mid-P method.14 To identify post–axi-cel risk factors that modulated infection density within the first 28 days, univariate and stepwise multivariate Poisson regressions were performed using an offset term defined as “days at risk.”
All statistical tests were conducted as 2-sided. Statistical significance was defined as P < .05. In the case of multiple comparisons, multiplicity adjusted P values were computed via the Holm-Šídák method.15 Analyses were performed using GraphPad Prism 8.0.2 (GraphPad Software, La Jolla, CA) and R version 3.6.1 using the survival package (v3.2-2).
Results
This study includes a consecutive series of 41 adult patients with R/R CD19+ LBCL who received axi-cel with minimum follow-up of 1 year for all durable responders. Patient and disease characteristics are shown in Table 1. Compared with the ZUMA-1 phase 2 trial cohort, the current study had more patients with non–germinal center B-cell–like disease (48.7%) and a history of primary refractory disease (46.3%). In addition, 16 (39%) patients treated would not have been eligible for inclusion in the ZUMA-1 trial, most often because of poor performance status, recent venous thromboembolism, or end-organ dysfunction. Despite these adverse features, therapeutic efficacy of axi-cel was similar, with an objective response rate of 87.8% and complete remission rate of 65.9% (supplemental Table 1). Eighteen (43.9%) patients received bridging therapy. Severe (grade ≥3) CRS occurred in 2.4% of patients, and severe ICANS occurred in 24.4%. A majority of patients received ≥1 dose of tocilizumab (82.9%) or corticosteroid (56.1%). The median duration of corticosteroid treatment of toxicities was 8 days (range, 1-30; supplemental Table 2). The median progression-free survival was 6.1 months, and the median OS was not reached after a median follow-up of 19.8 months (supplemental Figure 1). NRM was 2.4% (n = 1) at 1 year postinfusion, with the single death due to Pneumocystis jirovecii pneumonia (PJP) at 4.5 months postinfusion.
Hematologic recovery
Prior to LD chemotherapy, 46.3% of patients had an absolute lymphocyte count <500 cells per μL, 4.9% had an absolute neutrophil count (ANC) <1000 cells per μL, and 23.5% had serum IgG <400 mg/dL (supplemental Table 3). Following treatment with LD chemotherapy and axi-cel infusion, severe (grade 3 or 4) cytopenias occurred in 40 (97.6%) patients within the first 28 days (excluding lymphopenia as an expected effect of LD chemotherapy; Figure 1A). Accounting for censoring due to competing events, severe cytopenias persisted in 32 of 40 (80%) patients between days 29 and 180 postinfusion (Figure 1B), in 13 of 23 (56.5%) patients between days 181 and 365 postinfusion (Figure 1C), and in 6 of 17 (35.3%) patients between day 366 and last follow-up (Figure 1D). Patients whose severe cytopenias resolved to grade ≤2 within the first 28 days (36.6%; n = 15) had a monophasic recovery, with a mean duration of 14.6 ± 9.4 days; lymphopenia was often the last to resolve. Patients whose severe cytopenias persisted beyond the first 28 days (63.4%, n = 26) had more variable courses of recovery, with 8 (30.8%) patients experiencing a prolonged monophasic recovery with a mean duration of 91 ± 72 days; in these cases, lymphopenia was often the last to resolve. Overall, 26 (63.4%) patients received a packed red blood cell transfusion, and 14 (34.1%) patients received a platelet transfusion, with 34.6% and 42.9% of transfusions occurring beyond the first 28 days postinfusion, respectively. Growth factor (granulocyte colony-stimulating factor [G-CSF]) support was used in 21 (51.2%) patients with severe neutropenia beyond the first 28 days postinfusion. In those patients who experienced episodic courses of recovery, severe neutropenia was the most common recurrent cytopenia, with 5 (12.2%) patients experiencing recurrent episodes beyond 1 year postinfusion in ongoing remission (Figure 1E; supplemental Figure 2). The mean duration of grade ≥ 3 neutropenia in these patients was 80 ± 48 days. After accounting for censoring due to competing events, when comparing those patients with durable responses following axi-cel with those whose disease progressed or relapsed after axi-cel, there were no significant differences between the time courses of their hematologic recovery (supplemental Figure 3).
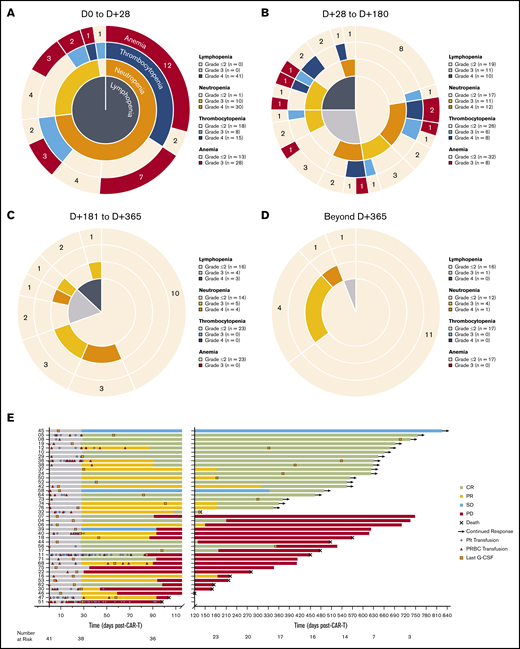
Hematologic recovery and transfusion utilization following CAR19. (A-D) Sunburst charts showing the overlapping prevalence of lymphopenia, neutropenia, thrombocytopenia, and anemia across 4 time periods post–axi-cel infusion. Concentric rings are organized in a hierarchical structure outward from the origin, with overlapping segments representing shared cytopenias within the same patient(s); the numbers shown in each outer ring segment represent patient(s) with that unique combination of cytopenias. The prevalence of individual cytopenias are shown in the accompanying keys. (E) Swimmer plot showing utilization of blood and platelet transfusions and time of last growth factor (G-CSF) administration for severe neutropenia. Patients are identified by study number on the left, and clinical response categorization is indicated by the bar color.
Hematologic recovery and transfusion utilization following CAR19. (A-D) Sunburst charts showing the overlapping prevalence of lymphopenia, neutropenia, thrombocytopenia, and anemia across 4 time periods post–axi-cel infusion. Concentric rings are organized in a hierarchical structure outward from the origin, with overlapping segments representing shared cytopenias within the same patient(s); the numbers shown in each outer ring segment represent patient(s) with that unique combination of cytopenias. The prevalence of individual cytopenias are shown in the accompanying keys. (E) Swimmer plot showing utilization of blood and platelet transfusions and time of last growth factor (G-CSF) administration for severe neutropenia. Patients are identified by study number on the left, and clinical response categorization is indicated by the bar color.
Cellular immune reconstitution
Circulating lymphocyte subsets were evaluated in peripheral blood following infusion of axi-cel using flow cytometry. Nontransduced (CAR−) CD4+ and CD8+ T cells were significantly depleted after the receipt of LD chemotherapy (CD4+ median, 31 cells per μL; interquartile range, 11-63; CD8+ median, 57 cells per μL; interquartile range, 4-160). Both populations recovered to significantly higher levels over the course of follow-up; however, the median CD4+ T-cell count by 18 months remained low at 150 cells per μL (range, 60-630). Accounting for censoring due to competing events, 12 of 22 (54.5%) patients had a CD4+ T-cell count <200 cells per μL by 6 months postinfusion, whereas 9 of 15 (60%) patients’ counts remained at <200 cells per μL at 1 year. CD8+ T cells recovered more rapidly than did CD4+ T cells, resulting in a CD4+/CD8+ ratio <1.0 in 10 of 15 (66.7%) patients by 18 months (Figure 2B-D). The duration of CD4+ T-cell count <200 cells per μL was significantly longer in older patients, but it was not influenced by the amount of prior therapy received, the receipt of bridging therapy, the severity of CRS, or the receipt of corticosteroids (supplemental Figure 6; supplemental Table 4). This protracted recovery of CD4+ T cells suggests that a majority of patients are at risk for opportunistic infection for ≥1 year following axi-cel.
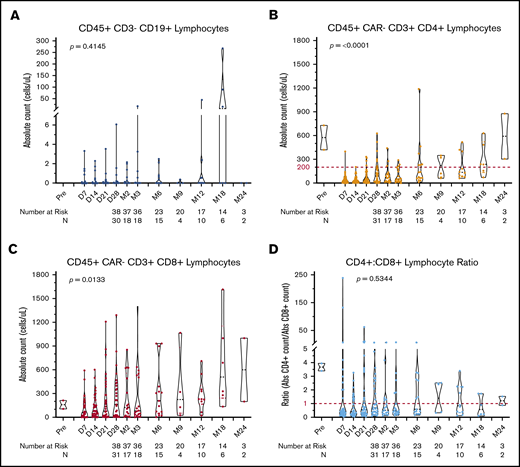
Lymphocyte subset recovery following CAR19. Violin plots showing the distribution of absolute cell counts for CD19+ B cells (A), CD4+ T cells (B), CD8+ T cells (C), and CD4/CD8 ratio measured by flow cytometry (D) performed on peripheral blood samples over the total duration of follow-up post–axi-cel infusion. The dashed red lines in B and D denote clinically actionable thresholds below which antimicrobial prophylaxis is used at the study center. The number of patients at risk, as well as the number of patients who underwent testing (N), is shown by time point below each plot. P values were obtained using the Kruskal-Wallis test; each time point was treated as an independent group.
Lymphocyte subset recovery following CAR19. Violin plots showing the distribution of absolute cell counts for CD19+ B cells (A), CD4+ T cells (B), CD8+ T cells (C), and CD4/CD8 ratio measured by flow cytometry (D) performed on peripheral blood samples over the total duration of follow-up post–axi-cel infusion. The dashed red lines in B and D denote clinically actionable thresholds below which antimicrobial prophylaxis is used at the study center. The number of patients at risk, as well as the number of patients who underwent testing (N), is shown by time point below each plot. P values were obtained using the Kruskal-Wallis test; each time point was treated as an independent group.
B-cell aplasia
To assess the on-target activity of axi-cel, we quantified CD19+ and/or CD22+ B cells in peripheral blood. B-cell aplasia occurred in 24 of 36 (66.7%) patients at the first assessment 7 days postinfusion (cohort range, 3-22; Figure 2A). Accounting for censoring due to competing events and sample availability, the number of patients with detectable B cells (loss of B-cell aplasia) by 6, 12, and 18 months postinfusion was 5 of 15 (33.3%), 4 of 10 (40%), and 3 of 6 (50%), respectively (Figure 3C). In total, 9 of 15 (60%) patients had ongoing B-cell aplasia at the time of last follow-up >12 months postinfusion (supplemental Figure 4). Patients with durable responses from axi-cel had significantly longer durations of B-cell aplasia than did those patients whose disease progressed or who relapsed after axi-cel (hazard ratio, 4.66; 95% CI, 1.76-12.29; Figure 3A). Conversely, patients who received bridging therapy or multiple doses of corticosteroids, both of which were previously identified as potential adverse prognostic factors in patients receiving axi-cel, had significantly shorter durations of B-cell aplasia (supplemental Table 4). Interestingly, loss of B-cell aplasia preceded or was concurrent with clinical or radiographic disease relapse or progression in 13 of 21 (61.9%) patients (median, 33 days prior; range 0-147). Of 21 patients whose disease relapsed or progressed, CD19 expression status on biopsy was available in 18 patients (85.7%). Within this cohort, there was no significant difference in the duration of B-cell aplasia between those whose lymphoma lost CD19 expression by immunohistochemistry or flow cytometry vs those who retained CD19 expression, although the number of evaluable patients for this analysis was small (Figure 3B).

B-cell aplasia and clinical response following CAR19. (A) Time-to-event analysis for recovery of CD19+ B cells detectable by flow cytometry performed on peripheral blood showed a significantly longer duration of B-cell aplasia in those patients who maintained a durable response to axi-cel. Patients were censored at the time of last follow-up; disease relapse or progression and death were considered competing events. Analysis was performed using the K-M method. (B) When patients who developed disease relapse or progression were stratified as CD19+ or CD19− status at relapse by immunohistochemistry or flow cytometry performed on lymphoma cells, there was no significant difference between the duration of B-cell aplasia observed. CD19 status at the time of relapse was available for 18 of 21 (85.7%) patients. (C) The proportion of patients with detectable B cells at each assessment time point, stratified by clinical response. The number of patients who underwent testing (N) is shown by time point below the plot. (D) Correlations between the duration of B-cell aplasia and the durations of other concurrent severe cytopenias; only noncensored durations from time-to-event analysis were included. Lines represent linear regression using least-squares fitting. P values shown were obtained by the likelihood ratio test computed using Cox proportional-hazards regression of the time-to-event variable vs the factor defining the 2 groups (A-B) and Spearman’s correlation analysis (D).
B-cell aplasia and clinical response following CAR19. (A) Time-to-event analysis for recovery of CD19+ B cells detectable by flow cytometry performed on peripheral blood showed a significantly longer duration of B-cell aplasia in those patients who maintained a durable response to axi-cel. Patients were censored at the time of last follow-up; disease relapse or progression and death were considered competing events. Analysis was performed using the K-M method. (B) When patients who developed disease relapse or progression were stratified as CD19+ or CD19− status at relapse by immunohistochemistry or flow cytometry performed on lymphoma cells, there was no significant difference between the duration of B-cell aplasia observed. CD19 status at the time of relapse was available for 18 of 21 (85.7%) patients. (C) The proportion of patients with detectable B cells at each assessment time point, stratified by clinical response. The number of patients who underwent testing (N) is shown by time point below the plot. (D) Correlations between the duration of B-cell aplasia and the durations of other concurrent severe cytopenias; only noncensored durations from time-to-event analysis were included. Lines represent linear regression using least-squares fitting. P values shown were obtained by the likelihood ratio test computed using Cox proportional-hazards regression of the time-to-event variable vs the factor defining the 2 groups (A-B) and Spearman’s correlation analysis (D).
The duration of all other severe cytopenias, including absolute lymphocyte count, ANC, platelet count, and CD4+ T-cell count, did not differ by clinical response to axi-cel (supplemental Figure 5; supplemental Table 4). The duration of CD4+ T-cell counts <200 cells per μL showed a trend toward significance when stratified by clinical response, whereas the duration of B-cell aplasia showed a trend toward significance when stratified by age (supplemental Figure 6). When comparing the duration of all severe cytopenias within the same patient, B-cell aplasia was strongly positively correlated only with the duration of the CD4+ T-cell count remaining <200 cells per μL (Figure 3D). This temporal association between B-cell and CD4+ T-cell recovery suggests that B-cell aplasia caused by axi-cel may directly contribute to prolonged CD4+ T-cell recovery.
Humoral immune reconstitution
Serum IgG levels decreased over the first 9 months following axi-cel infusion. In total, 23 of 37 (62.2%) patients had a serum IgG level < 400 mg/dL over the course of follow-up. Of those patients with normal serum IgG levels at baseline, new hypogammaglobulinemia occurred in 5 of 17 (29.4%) patients at a median of 2 months postinfusion. By 18 months, 8 of 18 (44.4%) patients’ IgG levels had not recovered to >400 mg/dL. Despite this prolonged recovery, only 15 (36.6%) patients received IVIG during follow-up after developing sinopulmonary infections. The median time to IVIG initiation was 160 days after axi-cel (range, 45-428). The incidence of hypogammaglobulinemia and IVIG use did not differ based on the number of prior lines of therapy received. Interestingly, patients who received IVIG maintained serum IgG levels that were similar to those in patients being followed expectantly (Figure 4A). In addition, we measured immunogenic antigen-specific IgG titers in 29 patients (Figure 4B-D). Although all titers decreased on average following axi-cel, the levels rarely fell below 50% of their pretreatment baseline, well above the minimum detectable limit. Those patients who received IVIG had higher average titers against each antigen than those who did not, although this effect was highly variable over the course of follow-up.

Serum immunoglobulin and antigen-specific antibody titers following CAR19. (A) Individual patient time courses showing recovery of serum IgG levels post–axi-cel infusion, stratified by whether patients received IVIG. The bold (Non-IVIG and IVIG) trend lines represents locally weighted scatterplot smoothing. Enzyme-linked immunosorbent assays were performed to detect antigen-specific IgG antibodies against Epstein-Barr virus (EBV) nuclear antigen 1 protein (EBNA1) (B), tetanus toxoid protein (C), and varicella-zoster virus (VZV) glycoprotein E (gpE) (D). Individual patient values relative to their pretreatment baseline are shown by the markers at each time point, with the bold (Non-IVIG and IVIG) trend lines connecting the geometric mean, stratified by whether patients received IVIG. P values shown were obtained by unpaired Student t tests between groups and Kruskal-Wallis tests within groups where each time point was treated as an independent group; both were adjusted for multiple comparisons using the Holm-Šídák method.
Serum immunoglobulin and antigen-specific antibody titers following CAR19. (A) Individual patient time courses showing recovery of serum IgG levels post–axi-cel infusion, stratified by whether patients received IVIG. The bold (Non-IVIG and IVIG) trend lines represents locally weighted scatterplot smoothing. Enzyme-linked immunosorbent assays were performed to detect antigen-specific IgG antibodies against Epstein-Barr virus (EBV) nuclear antigen 1 protein (EBNA1) (B), tetanus toxoid protein (C), and varicella-zoster virus (VZV) glycoprotein E (gpE) (D). Individual patient values relative to their pretreatment baseline are shown by the markers at each time point, with the bold (Non-IVIG and IVIG) trend lines connecting the geometric mean, stratified by whether patients received IVIG. P values shown were obtained by unpaired Student t tests between groups and Kruskal-Wallis tests within groups where each time point was treated as an independent group; both were adjusted for multiple comparisons using the Holm-Šídák method.
Infectious complications
There were 3 (7.3%) patients who had an infection in the interval between apheresis and LD chemotherapy; in 2 patients this developed after the receipt of chemoimmunotherapy for bridging treatment. These included Candida parapsilosis pyelonephritis, respiratory syncytial virus bronchiolitis, and rhinovirus upper respiratory tract infection. In addition, 2 (4.9%) patients developed mucocutaneous candidiasis after the receipt of LD chemotherapy. In the first 28 days following axi-cel infusion, 19 (46.3%) patients had an infection, with the majority being mild to moderate in severity (13/19; 68.4%). The most common etiology was viral respiratory tract infections (21.1%), followed by Clostridioides difficile infections (15.8%; supplemental Table 5). There was 1 severe infection due to HHV-6 viral meningoencephalitis requiring intensive care unit admission that occurred in the first 28 days. By 28 days, the cumulative incidence of first infection estimated by the K-M method was 55.2% (95% CI, 37.4-69.8; viral, 19.7%; bacterial, 16.3%; fungal, 8.9%), by 6 months it was 69% (95% CI, 55.4-79.2; viral, 23.7%; bacterial, 30.1%; fungal, 8.9%), and by 12 months it was 89.7% (95% CI, 83.9-93.4; viral, 56.4%; bacterial, 50.6%; fungal, 8.9%) (Figure 5A). The calculated incidence rate of infection, or infection density, within the first 28 days was 2.35 infections per 100 days at risk. This was significantly higher than at any other time period during follow-up when controlling for competing events (IRR, 6.10; 95% CI, 3.76-9.82). The same pattern was seen with severe infections as well (Figure 5B; supplemental Table 6). In a univariate Poisson regression model, the development of grade 2 CRS or ICANS or receipt of corticosteroids were each associated with an increased infection density. When these factors were combined in a stepwise multivariate model, only receipt of corticosteroids remained significantly associated with increased infection density (hazard ratio, 3.69; 95% CI, 1.18-16.5) (supplemental Table 7). Of note, all cases of cytomegalovirus infection (n = 4) occurred within 2 weeks of initiating corticosteroids. In each case, patients developed viremia without evidence of end-organ involvement, and only 1 patient required antiviral therapy after tapering corticosteroids completely.
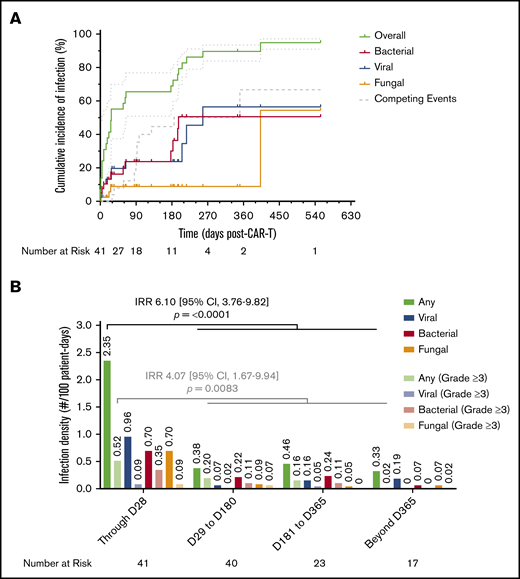
Cumulative incidence of infection and infection density following CAR19. (A) Cumulative incidence time-to-event analysis among the total cohort (N = 41) showing overall time to first infection of any type, as well as subsets for bacterial, viral, and fungal infections, over the total duration of follow-up after axi-cel infusion. Patients were censored at the time of last follow-up; disease relapse or progression and death were defined as competing events. Dotted lines represent 95% CI. (B) Infection densities are shown for any type and severity, as well as subsets for bacterial, viral, and fungal infections of any severity, occurring during 4 time periods following axi-cel infusion. The same groupings are interleaved for severe (CTCAE grade ≥3) infections alone. Calculated infection densities are shown above each bar. P values shown were obtained by calculating an IRR between the first 28 days of follow-up and the remainder of follow-up using the mid-P method and comparing using Fisher’s exact test.
Cumulative incidence of infection and infection density following CAR19. (A) Cumulative incidence time-to-event analysis among the total cohort (N = 41) showing overall time to first infection of any type, as well as subsets for bacterial, viral, and fungal infections, over the total duration of follow-up after axi-cel infusion. Patients were censored at the time of last follow-up; disease relapse or progression and death were defined as competing events. Dotted lines represent 95% CI. (B) Infection densities are shown for any type and severity, as well as subsets for bacterial, viral, and fungal infections of any severity, occurring during 4 time periods following axi-cel infusion. The same groupings are interleaved for severe (CTCAE grade ≥3) infections alone. Calculated infection densities are shown above each bar. P values shown were obtained by calculating an IRR between the first 28 days of follow-up and the remainder of follow-up using the mid-P method and comparing using Fisher’s exact test.
Following day 28, the majority of infections occurred after 6 months postinfusion and consisted primarily of viral upper respiratory tract infections (23.5%), bacterial pneumonias (32.4%), and herpes zoster (20.6%; supplemental Table 5). In those patients who experienced grade ≥3 neutropenia after day 28, the incidence of febrile neutropenia was rare (5%), with 5 events occurring in 4 patients out of 101 collective days with an ANC <1000 cells per μL among 23 patients. Nearly all of events (80%) were respiratory infections (2 viral, 1 PJP, 1 unidentified), with 3 classified as severe. In total, there were 7 severe infections requiring intensive care unit admission in 5 patients that occurred after day 28, including 3 cases of bacterial pneumonia (1 complicated by Streptococcus pneumoniae bacteremia), 1 case of diverticulitis complicated by pericolonic abscess, and 3 cases of PJP. Each case of PJP infection occurred in patients who previously achieved durable responses to axi-cel therapy and within 3 months of completing a prespecified duration of trimethoprim-sulfamethoxazole (TMP-SMX) prophylaxis. All patients developed severe disease requiring intubation and mechanical ventilation and were treated with 15 mg/kg per day of TMP-SMX and high-dose prednisone (>60 mg/d). In each case, their CD4+ T-cell count was <200 cells per μL. One patient died from complications related to PJP infection at 4 months postinfusion, which represented the only death unrelated to lymphoma within this study.
We evaluated several facets of immune reconstitution to determine whether any were associated with risk of infection after day 28. When comparing patients based on the amount of prior lymphoma-directed therapy, whether they attained a durable clinical response to axi-cel, required prolonged G-CSF support beyond day 28, or required IVIG support, only those patients with >3 lines of prior therapy had a significant increase in their infection density over the duration of follow-up (IRR, 1.81; 95% CI, 1.13-2.91) (supplemental Table 8). A landmark analysis of CD4+ T-cell count recovery by 6 months, split into those whose count recovered to >200 cells per μL (n = 10) and those whose count remained <200 cells per μL (n = 12), did not show any significant differences in the cumulative infection density over any time period and showed similar trajectories of time to first infection, regardless of severity (supplemental Table 5).
Discussion
This single-institution retrospective analysis of 41 LBCL patients receiving axi-cel reports the incidence of hematologic toxicity, immune suppression, and infectious risks in short-term and long-term follow-up. These interrelated toxicities represent an important source of posttreatment morbidity and NRM, although to date there are no evidence-based guidelines addressing supportive care measures to reduce infection risk post–CAR T-cell therapy.
Although the vast majority of severe cytopenias resolved completely within the first 90 days, nearly two thirds of patients in the current cohort required transfusion support with red blood cells, platelets, or both during the initial post–axi-cel period. Furthermore, half of the patients required G-CSF support to maintain ANC >1000 cells per μL, and one third received IVIG support. Our analysis of patients with recurrent neutropenia showed that this finding was not associated with excess infections compared with sustained count recovery. Similarly, the prevalence of hypogammaglobulinemia due to the on-target B-cell ablation from axi-cel was observed in a significant proportion of the cohort, yet it was not associated with excess infections compared with higher IgG levels. Despite the delayed immune reconstitution present in a majority of patients, the NRM in our cohort compared favorably with contemporary reports of autologous hematopoietic stem cell transplant, suggesting a benefit to supportive care measures.17,18
Episodic severe neutropenia represented the most common hematologic toxicity observed during the long-term follow-up of our cohort, similar to prior reports.1,5-8 The exact mechanism of myelosuppression in the post-CAR19 setting is unknown, but it does not appear to be due to bone marrow dysfunction or damage (eg, myelodysplastic syndromes) in the majority of patients.19,20 Delayed persistent neutropenia has been reported following other B-cell–directed therapies, including rituximab and blinatumomab.21-23 Prior mechanistic studies, including in the post-CAR19 setting, have suggested that posttreatment surges in growth and retention factors driving B-cell lymphopoiesis, including B-cell-activating factor (BAFF) and stromal-derived factor-1, lead to attenuation of myelopoiesis and neutrophil egress from the bone marrow.7,23,24 In 1 study, a dose-response relationship between levels of BAFF and late-onset neutropenia was noted, suggesting that more pronounced B-cell depletion may predispose to an increased risk for competitive suppression of myelopoiesis.24 Further studies are needed to assess the temporal relationship between these serologic markers and lymphocyte subset recovery following CAR19.
This study recapitulated recently published observations demonstrating the prolonged delay in CD4+ T-cell recovery following axi-cel, with ≥50% of patients having counts <200 cells per μL at 1 year.5,6 This extended period of risk for opportunistic infections represents a significantly underrecognized phenomenon following CAR19. A similar prolonged trajectory of CD4+ T-cell recovery has been shown after intensive chemotherapy in solid tumors25-27 and lymphoid malignancies.28,29 Accompanying mechanistic studies have suggested that this delayed CD4+ T-cell recovery following intensive chemotherapy occurs predominantly as a result of inefficient thymus-independent peripheral T-cell expansion.30-32 However, the chemotherapy dose intensity received by patients in those early studies is, on average, 4 to 8 times higher than the LD chemotherapy regimen received pre-CAR19. In regimens in which the dose intensity of fludarabine and cyclophosphamide (FC) are more closely matched (eg, FC or FC plus rituximab [FCR]), CD4+ T-cell counts recover to significantly higher levels by 1 year posttreatment compared with those observed in our cohort.33-35 The significant association between older age and prolonged CD4+ T-cell depletion seen in our cohort could represent a demographic proxy for thymic function. However, the lack of association between age and the duration of B-cell aplasia, despite the latter correlating strongly with the duration of CD4+ T-cell depletion, suggests that chemotherapy dose-dependent myelotoxicity and thymic function are not the sole mechanistic explanations for this observed phenomenon following axi-cel.
In our study, the recovery in CD4+ T-cell count showed a strong positive correlation with time to recovery of B cells, recapitulating a similar temporal relationship demonstrated in patients receiving B-cell–directed therapy with rituximab.36-38 Prior animal models have shown that depletion of CD19+ or CD20+ B cells in lymph nodes leads to impairment in major histocompatibility complex II–dependent CD4+ T-cell activation and expansion.39 Conversely, antigen-specific humoral immunity against vaccination targets appeared minimally affected, suggesting that CD19− plasma cells continue to function despite the loss of other B-cell subsets.40,41 Taken together, these findings suggest that prolonged immune reconstitution, especially CD4+ T-cell recovery, may be mechanistically linked to the on-target B-cell depletion achieved with effective CAR19 and have similar implications for alternative B-cell antigen targets, such as BAFF.
Interestingly, we observed an association between the duration of B-cell aplasia and clinical response to axi-cel. Although this has been observed in patients with B-cell acute lymphoblastic leukemia undergoing CAR19, it has not been reported in LBCL.42,43 A majority of patients had early loss of B-cell aplasia precede clinical disease relapse, which did not differ based on CD19 status at the time of relapse. Although antigen loss occurs in ∼33% of patients with LBCL receiving CAR19, in our cohort this was not a significant factor affecting the duration of B-cell aplasia following axi-cel.4,44 The clear association between durable B-cell aplasia and clinical response, as well as the increased prevalence of early B-cell recovery in patients with disease relapse or progression, suggests that assessment of peripheral B cells might serve as an effective proxy for ongoing CAR19 activity in the initial months following axi-cel. However, the protracted duration of B-cell aplasia seen in our cohort and others suggests that extensive on-target ablation by successful CAR19 may result in lasting damage to the B-cell niche well beyond CAR T-cell persistence.45
Recent studies have shown that LBCL survivors have significant ongoing risks for a variety of infections outside of CAR19.46 Despite similar overall infection densities, our data highlight several patients who developed severe opportunistic infections after discontinuing antimicrobial prophylaxis before recovery of cellular or humoral immunity.6,8 The timing of these infectious events guided modification of institutional antimicrobial prophylaxis guidelines over the course of the study (Table 2). After noting episodes of herpes zoster coinciding with the cessation of antiviral therapy, acyclovir prophylaxis was changed to 800 mg twice a day to improve compliance and was iteratively extended to 18 months.47 Similarly, the surge in viral and bacterial respiratory tract infections after 6 months coincided with a serum IgG nadir and led to subsequent initiation of IVIG for many patients. In the case of PJP prophylaxis, 2 patients experienced severe (and in 1 case fatal) infections within 1 month of discontinuing TMP-SMX prophylaxis at 3 months, which led to the extension of prophylaxis to 1 year. An additional case developed in a patient at ∼15 months postinfusion after discontinuing TMP-SMX prophylaxis, at which time all 3 patients were noted to have CD4+ T-cell counts <200 cells per μL, which led to the extension of prophylaxis beyond 12 months with regular monitoring of CD4+ T-cell counts.48,49 As other centers reported cases of viral hepatitis B reactivation and invasive fungal respiratory infections, these were incorporated into our guidelines.5,8
Because CAR19s, such as axi-cel, continue to produce a dramatic shift in the survival of patients with R/R LBCL, the need for comprehensive supportive care emphasized by these data will continue to increase in importance. Severe infections remain a low risk for patients with durable responses to CAR19; however, the impact of ongoing myelosuppression and poor immune reconstitution on patients with disease relapse who require additional lines of therapy remains unclear. Because the current study is limited by its retrospective nature and cohort size, further studies are needed to identify modifiable risk factors for infection post-CAR19. In lieu of comprehensive risk stratification, randomized trials comparing prophylaxis strategies, or other robust data on interventions to mitigate infectious risks, we have implemented best practice guidelines for short- and long-term monitoring and antimicrobial prophylaxis.
Deidentified individual participant data that underlie the reported results will be made available upon request. Proposals for access should be sent to David B. Miklos (dmiklos@stanford.edu).
Acknowledgments
The authors thank the staff and faculty of the Stanford University Blood and Marrow Transplantation Program and the Stanford Center for Cancer Cell Therapy for tireless work caring for the patients involved in this study and Fang Wu for assistance with assay development and data acquisition.
This work was supported by National Institutes of Health/National Cancer Institute grants 2P01CA049605-29A1 (C.L.M. and D.M.) and 5P30CA124435 (C.L.M.), and by the Virginia and D. K. Ludwig Fund for Cancer Research. C.L.M. is a member of the Parker Institute for Cancer Immunotherapy, which supports the Stanford University Cancer Immunotherapy Program.
Authorship
Contribution: J.H.B., D.J.E., D.B.M., and S.S. designed the study and interpreted the data; J.H.B., J.S.T., and J.Y.S. analyzed the data and created the figures; J.H.B., D.B.M., and S.S. wrote the manuscript; and all authors contributed to data collection, writing and revision of the manuscript, and approved the final version.
Conflict-of-interest disclosure: P.S. has received research support from Kite Pharma-Gilead. A.R.R. has received research support from Pharmacyclics/AbbVie; has served on ad hoc scientific advisory boards for Nohla Therapeutics and Kaleido; has served as an expert witness for US Department of Justice; and his brother works for Johnson & Johnson. T.L. has been a member of the speaker’s bureau for Kite Pharma-Gilead. C.L.M. has acted as a consultant for Lyell, NeoimmuneTech, Nektar Therapeutics, and Apricity Therapeutics; has received royalties from the National Institutes of Heath and Juno Therapeutics for CD22-CARl and has equity in Lyell and Allogene Therapeutics. D.B.M. has acted as a consulting for Kite Pharma-Gilead, Juno Therapeutics-Celgene, Novartis, Janssen Pharmaceuticals, and Pharmacyclics and has received research support from Kite Pharma-Gilead, Allogene Therapeutics, Pharmacyclics, Miltenyi Biotec, and Adaptive Biotechnologies. S.S. has acted as a consultant for Janssen Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: David B. Miklos, Stanford University, 269 W Campus Dr, CCSR 2205, Stanford, CA 94305; e-mail: dmiklos@stanford.edu; and Surbhi Sidana, Stanford University, 300 Pasteur Dr, Room H0101C, Stanford, CA 94305; e-mail: surbhi.sidana@stanford.edu.



