von Willebrand disease (VWD) is the most common inherited bleeding disorder known in humans. Accurate and timely diagnosis presents numerous challenges.
These evidence-based guidelines of the American Society of Hematology (ASH), the International Society on Thrombosis and Haemostasis (ISTH), the National Hemophilia Foundation (NHF), and the World Federation of Hemophilia (WFH) are intended to support patients, clinicians, and other health care professionals in their decisions about VWD diagnosis.
ASH, ISTH, NHF, and WFH established a multidisciplinary guideline panel that included 4 patient representatives and was balanced to minimize potential bias from conflicts of interest. The Outcomes and Implementation Research Unit at the University of Kansas Medical Center (KUMC) supported the guideline-development process, including performing or updating systematic evidence reviews up to 8 January 2020. The panel prioritized clinical questions and outcomes according to their importance for clinicians and patients. The panel used the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach, including GRADE Evidence-to-Decision frameworks, to assess evidence and make recommendations, which were subsequently subject to public comment.
The panel agreed on 11 recommendations.
Key recommendations of these guidelines include the role of bleeding-assessment tools in the assessment of patients suspected of VWD, diagnostic assays and laboratory cutoffs for type 1 and type 2 VWD, how to approach a type 1 VWD patient with normalized levels over time, and the role of genetic testing vs phenotypic assays for types 2B and 2N. Future critical research priorities are also identified.
Summary of recommendations
These guidelines are based on updated and original systematic reviews of evidence conducted under the direction of the Outcomes and Implementation Research Unit at the University of Kansas Medical Center (KUMC). The panel followed best practices for guideline development recommended by the Institute of Medicine and the Guidelines International Network (G-I-N).1-3 The panel used the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach4-10 to assess the certainty in the evidence and formulate recommendations.
von Willebrand disease (VWD) is a common, inherited bleeding disorder. The current classification includes types 1 and 3, which are characterized by quantitative deficiencies of von Willebrand factor (VWF), as well as types 2A, 2B, 2M, and 2N, which are qualitative variants. Clinically, VWD patients experience excessive mucocutaneous bleeding, including heavy menstrual bleeding, epistaxis, easy bruising, prolonged bleeding from minor wounds and the oral cavity, and gastrointestinal bleeding, as well as bleeding after dental work, childbirth, and surgery, with musculoskeletal bleeding also seen in the most severe cases. Treatment includes adjunctive therapies, such as tranexamic acid, and therapies that directly increase the levels of VWF, such as desmopressin and VWF concentrates. The accurate and timely diagnosis of VWD remains a challenge for clinicians and patients.
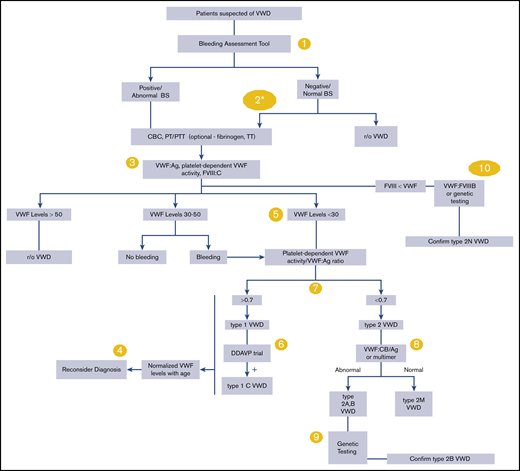
Please see Figure 1 for an overall algorithm addressing the diagnosis of VWD.
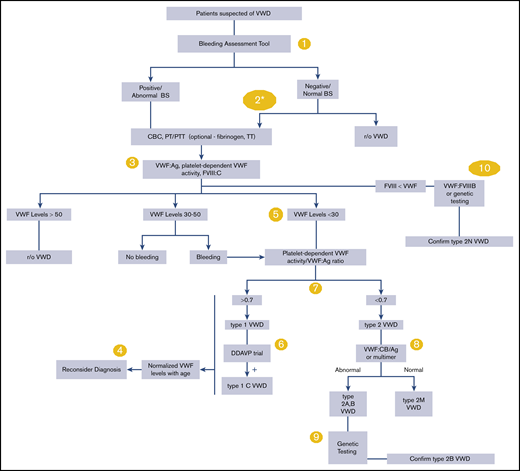
An overall algorithm addressing the diagnosis of VWD. The numbers in the yellow circles correspond to guideline questions. VWF levels refer to VWF antigen (VWF:Ag) and/or platelet-dependent VWF activity. The algorithm says VWF level 30 to 50 for simplicity; this refers to VWF levels of 0.30 to 0.50 IU/mL, with the caveat that the lower limit of the normal range as determined by the local laboratory should be used if it is <0.50 IU/mL. *Men and children, referred to a hematologist and/or first-degree relative affected with VWD. BS, bleeding score; CBC, complete blood count; DDAVP, desmopressin; FVIII, factor FVIII; FVIII:C, FVIII coagulant activity; PT, prothrombin time; PTT, partial thromboplastin time; r/o, rule out; TT, thrombin time; VWF:CB/Ag, ratio of VWF collagen binding to antigen; VWF:FVIIIB, VWF FVIII binding.
An overall algorithm addressing the diagnosis of VWD. The numbers in the yellow circles correspond to guideline questions. VWF levels refer to VWF antigen (VWF:Ag) and/or platelet-dependent VWF activity. The algorithm says VWF level 30 to 50 for simplicity; this refers to VWF levels of 0.30 to 0.50 IU/mL, with the caveat that the lower limit of the normal range as determined by the local laboratory should be used if it is <0.50 IU/mL. *Men and children, referred to a hematologist and/or first-degree relative affected with VWD. BS, bleeding score; CBC, complete blood count; DDAVP, desmopressin; FVIII, factor FVIII; FVIII:C, FVIII coagulant activity; PT, prothrombin time; PTT, partial thromboplastin time; r/o, rule out; TT, thrombin time; VWF:CB/Ag, ratio of VWF collagen binding to antigen; VWF:FVIIIB, VWF FVIII binding.
Interpretation of strong and conditional recommendations
The strength of a recommendation is expressed as either strong (“the guideline panel recommends...”), or conditional (“the guideline panel suggests…”) and has the following interpretation:
Strong recommendation
For patients: Most individuals in this situation would want the recommended course of action, and only a small proportion would not.
For clinicians: Most individuals should follow the recommended course of action. Formal decision aids are not likely to be needed to help individual patients make decisions consistent with their values and preferences.
For policy makers: The recommendation can be adopted as policy in most situations. Adherence to this recommendation according to the guideline could be used as a quality criterion or performance indicator.
For researchers: The recommendation is supported by credible research or other convincing judgments that make additional research unlikely to alter the recommendation. On occasion, a strong recommendation is based on low or very low certainty in the evidence. In such instances, further research may provide important information that alters the recommendations.
Conditional recommendation
For patients: The majority of individuals in this situation would want the suggested course of action, but many would not. Decision aids may be useful in helping patients to make decisions consistent with their individual risks, values, and preferences.
For clinicians: Recognize that different choices will be appropriate for individual patients and that you must help each patient arrive at a management decision consistent with their values and preferences. Decision aids may be useful in helping individuals to make decisions consistent with their individual risks, values, and preferences.
For policy makers: Policy making will require substantial debate and involvement of various stakeholders. Performance measures about the suggested course of action should focus on whether an appropriate decision-making process is duly documented.
For researchers: This recommendation is likely to be strengthened (for future updates or adaptation) by additional research. An evaluation of the conditions and criteria (and the related judgments, research evidence, and additional considerations) that determined the conditional (rather than strong) recommendation will help identify possible research gaps.
Interpretation of good practice statements
As described by the GRADE Guidance Group, good practice statements endorse interventions or practices that the guideline panel agreed have unequivocal net benefit yet may not be widely recognized or used.11 Good practice statements in these guidelines are not based on a systematic review of available evidence. Nevertheless, they may be interpreted as strong recommendations.
Recommendations
Bleeding-assessment tools.
RECOMMENDATION 1.
For patients with a low probability of VWD (eg, seen in the primary care setting), the panel recommends using a validated bleeding-assessment tool (BAT) as an initial screening test to determine who needs specific blood testing over nonstandardized clinical assessment (strong recommendation based on moderate certainty in the evidence from diagnostic accuracy studies ⊕⊕⊕◯).
Remarks:
This recommendation applies predominantly to adult women, as the data supporting the use of a BAT as a screening tool is strongest in this patient group.
The quality of nonstandardized clinical assessment will vary among the users of these guidelines.
Specific blood testing for VWD refers to VWF antigen (VWF:Ag), platelet-dependent VWF activity (eg, VWF glycoprotein IbM [VWF:GPIbM]), and factor VIII (FVIII) coagulant activity (FVIII:C).
RECOMMENDATION 2.
For patients with an intermediate probability of VWD (eg, referred to a hematologist), the panel suggests against relying on a BAT to decide whether to order specific blood testing (conditional recommendation based on moderate certainty in the evidence from diagnostic accuracy studies ⊕⊕⊕◯).
Remarks:
This recommendation addresses patients with an intermediate VWD pretest probability (∼20%) corresponding to those typically referred for hematology evaluation because of an abnormal personal bleeding history or abnormal initial laboratory tests (eg, prolonged activated partial thromboplastin time [aPTT]) (including men and children).
Beyond their utility as a screening test in the primary care setting, BATs can be used in the referral setting to assess and document the severity of bleeding and can be used in conjunction with specific blood testing as part of the initial diagnostic approach.
Specific blood testing for VWD refers to VWF:Ag, platelet-dependent VWF activity (eg, VWF:GPIbM), and FVIII:C.
RECOMMENDATION 3.
For patients with a high probability of VWD (eg, affected first-degree relative), the panel recommends against relying on a BAT to decide whether to order specific blood testing (strong recommendation based on moderate certainty in the evidence from diagnostic accuracy studies ⊕⊕⊕◯).
Remarks:
This recommendation addresses patients with a high VWD pretest probability (∼50%) corresponding to those typically referred for hematology evaluation because of an affected first-degree relative regardless of their bleeding symptoms or initial laboratory tests (including men and children).
Beyond their utility as a screening test in the primary care setting, BATs can be used in the referral setting to assess and document the severity of bleeding and can be used in conjunction with specific blood testing as part of the initial diagnostic approach.
Specific blood tests for VWD refer to VWF:Ag, platelet-dependent VWF activity (eg, VWF:GPIbM), and FVIII:C.
Assays of platelet-binding activity of VWF.
RECOMMENDATION 4.
The panel suggests newer assays that measure the platelet-binding activity of VWF (eg, VWF:GPIbM, VWF:GPIbR) over the VWF ristocetin cofactor assay (VWF:RCo) (automated or nonautomated assay) for the diagnosis of VWD (conditional recommendation based on low certainty in the evidence from diagnostic accuracy studies ⊕⊕◯◯).
GOOD PRACTICE STATEMENT.
VWF activity assays should be performed in a laboratory with appropriate expertise.
VWF levels that normalize with age.
RECOMMENDATION 5.
The panel suggests reconsidering the diagnosis as opposed to removing the diagnosis for patients with previously confirmed type 1 VWD who now have VWF levels that have normalized with age (conditional recommendation based on very low certainty in the evidence of effects ⊕◯◯◯).
Remarks:
With this recommendation, the panel worked under the assumption that the original diagnosis of type 1 VWD was accurate.
Aging and comorbidities are known to increase VWF levels. However, the association between the increased VWF levels and bleeding symptoms is not established.
Decisions about reconsidering or removing the diagnosis should consider the patient’s values and preferences and be informed by a shared decision-making process.
Type 1 VWD.
RECOMMENDATION 6.
The panel recommends a VWF level of <0.30 IU/mL regardless of bleeding, and for patients with abnormal bleeding, a VWF level of <0.50 IU/mL to confirm the diagnosis of type 1 VWD (strong recommendation based on low certainty in the evidence of effects ⊕⊕◯◯).
Remarks:
VWF level(s) refers to VWF:Ag and/or platelet-dependent VWF activity (eg, VWF:GPIbM).
The lower limit of the normal range as determined by the local laboratory should be used if it is <0.50 IU/mL. ABO-specific reference ranges are not required.
VWF is an acute-phase reactant that increases in response to a variety of stimuli (eg, bleed, trauma, pregnancy). VWD diagnostic testing should be performed when patients are at a baseline state of health.
Type 1C VWD.
RECOMMENDATION 7.
The panel suggests against using VWF propeptide (VWFpp)/VWF:Ag (the ratio of VWF propeptide to antigen) and rather using a desmopressin trial with 1- and 4-hour postinfusion blood work to confirm increased VWF clearance for patients with VWD suspected of type 1C (conditional recommendation based on low certainty in the evidence from diagnostic accuracy studies ⊕⊕◯◯).
Type 2 VWD.
RECOMMENDATION 8.
The panel suggests against a platelet-dependent VWF activity/VWF:Ag ratio <0.5 cutoff, and rather using a higher cutoff of <0.7 to confirm type 2 VWD (2A, 2B, or 2M) for patients with an abnormal initial VWD screen (conditional recommendation based on very low certainty in the evidence from diagnostic studies ⊕◯◯◯).
Remark:
Some patients with type 2 VWD have normal VWF:Ag and platelet-dependent VWF activity but a low ratio of platelet-dependent VWF activity/VWF:Ag.
RECOMMENDATION 9.
The panel suggests either VWF multimer analysis or VWF collagen binding (VWF:CB)/VWF:Ag (the ratio of VWF collagen binding to antigen) to diagnose type 2 VWD for patients suspected of type 2A, 2B, or 2M in need of additional testing (conditional recommendation based on very low certainty in the evidence from diagnostic accuracy studies ⊕◯◯◯).
Remark:
Most laboratories that do the VWF:CB assay use type I and/or III collagen, which is known to be a surrogate for the presence of high-molecular-weight VWF.
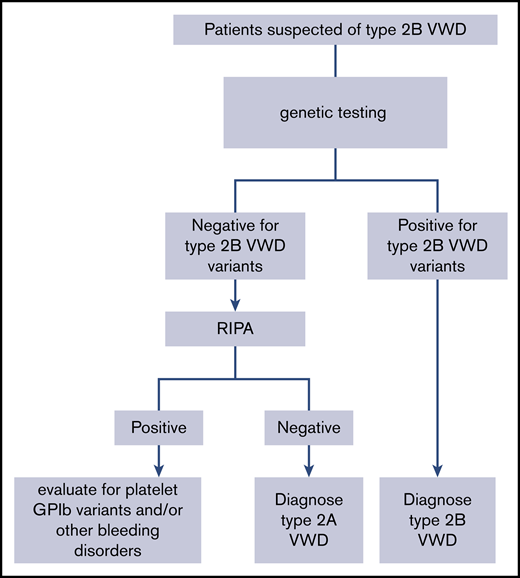
RECOMMENDATION 10.
The panel suggests targeted genetic testing over low-dose ristocetin-induced platelet agglutination (RIPA) to diagnose type 2B VWD for patients suspected of type 2A or 2B in need of additional testing (Figure 2) (conditional recommendation based on low certainty in the evidence from diagnostic accuracy studies ⊕⊕◯◯).
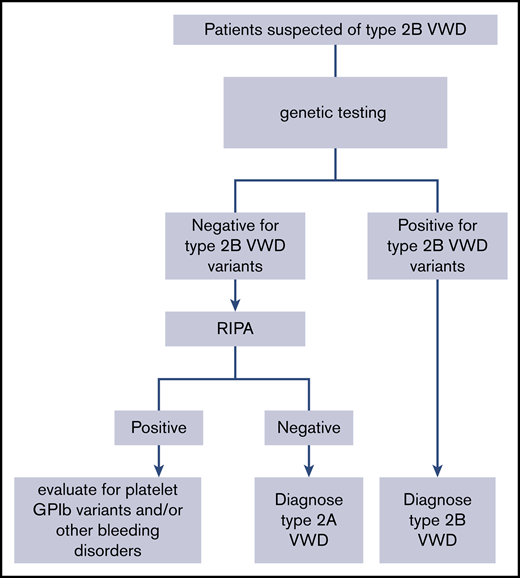
An algorithm for the diagnosis of type 2B VWD. GPIb, glycoprotein Ib; RIPA, ristocetin-induced platelet agglutination.
An algorithm for the diagnosis of type 2B VWD. GPIb, glycoprotein Ib; RIPA, ristocetin-induced platelet agglutination.
RECOMMENDATION 11.
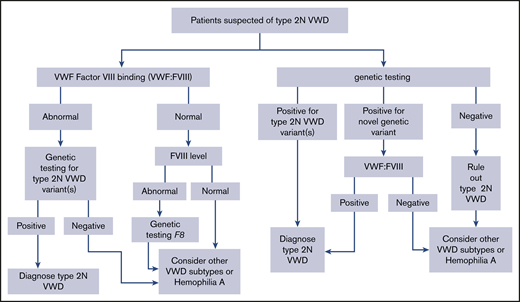
The panel suggests using either VWF FVIII binding (VWF:FVIIIB) or targeted genetic testing (when available) for patients with suspected type 2N VWD in need of additional testing (Figure 3) (conditional recommendation based on low certainty in the evidence from diagnostic accuracy studies ⊕⊕◯◯).
Values and preferences
These recommendations place the highest value on not missing the diagnosis in affected patients in order to ensure access to care. The panel considered the following outcomes as critical for clinical decision-making across questions: major bleeding, transfusion and treatment, gastrointestinal bleeding, blood loss, symptom severity, minor bleeding, mortality, and unnecessary testing. These outcomes will be affected by the accurate diagnosis of different subtypes of VWD and avoiding inaccurate mislabeling of patients.
Explanations and other considerations
These recommendations take into consideration cost and cost-effectiveness, resource requirements, impact on health equity, acceptability, and feasibility. Many included studies suffered from a high risk of bias due to the lack of clear reference standards and issues with patient selection.
Introduction
Aims of these guidelines and specific objectives
The purpose of these guidelines is to provide evidence-based recommendations on the diagnosis of VWD. The primary goals of these guidelines are to review, critically appraise, and implement evidence-based recommendations that will improve the accurate identification of affected patients while minimizing inappropriate testing and the harms of overdiagnosis. Through improved provider and patient education using the available evidence and evidence-based recommendations, these guidelines aim to provide clinical decision support for shared decision-making that will result in accurate VWD diagnoses, which will lead to improved education and counseling of patients as well as effective treatment and prevention of bleeding episodes. The target audience includes hematologists, general practitioners, internists, other clinicians and decision-makers, and patients. Policy makers who may be interested in these guidelines include those involved in developing local, national, or international plans with the goal of improving the lives of patients living with VWD. This document may also serve as the basis for adaptation by local, regional, or national guideline panels.
Description of the health problem
In 1926, a Finnish physician, Erik von Willebrand, published a description of a new bleeding disorder that he observed in a family living in the Åland Islands in the Baltic Sea.12 The index case was a young woman who bled to death at the time of her fourth menstrual period; many other family members also suffered from excessive bleeding. In the original report, the disease was referred to as “pseudohemophilia”; however, it later came to be known as VWD. VWD is caused by deficiency or dysfunction of the multimeric glycoprotein VWF, which plays key hemostatic roles in the circulation, including platelet adhesion and aggregation at sites of vascular injury, and acts as a chaperone for FVIII.13 The VWF gene is located on the long arm of chromosome 12 and comprises 52 exons that encode 2813 amino acids.14
VWD is characterized by excessive mucocutaneous bleeding, such as heavy menstrual bleeding, epistaxis, easy bruising, prolonged bleeding from minor wounds and the oral cavity, and gastrointestinal bleeding, as well as bleeding after dental work, childbirth, and surgery, with musculoskeletal bleeding, including joint bleeding seen in more severe cases.13 It is the most common bleeding disorder known in humans and is inherited equally between men and women; however, women are more likely to come to medical attention because of gynecologic and obstetric bleeding. VWD prevalence estimates range from ∼1 in 100 to 1 in 10 000.13,15-17 At the level of primary care, ∼1 in 1000 individuals are affected and require medical attention for bleeding.18,19 The current International Society on Thrombosis and Haemostasis (ISTH) classification recognizes 3 types: type 1 is a partial quantitative deficiency of VWF, type 2 is caused by qualitative abnormalities of VWF, and type 3 is a virtual absence of the VWF protein with associated very low FVIII levels. Type 2 VWD is further divided into 4 subtypes: type 2A is characterized by reduced or absent high-molecular-weight VWF, type 2B results from a gain of function in VWF that increases its affinity for platelets, type 2M is caused by reduced VWF interactions with platelets or collagen, and type 2N results from reduced binding of VWF to FVIII.20 The panel approached this guideline within the framework of this classification system with the addition of type 1C VWD, which is caused by increased VWF clearance, given that it has management implications for patients.21,22
A major challenge for affected patients is achieving an accurate and timely diagnosis.23-26 Patients experience delays of 15 years or more from the onset of bleeding symptoms to a VWD diagnosis, and confusion remains about the distinction and importance of the types and subtypes described above.27 Barriers to an accurate diagnosis include a lack of understanding of the difference between normal and abnormal bleeding symptoms, lack of clarity around an appropriate diagnostic approach, and limited availability and expertise of specialized laboratory testing. These considerations informed the panel’s deliberations, with a high value being placed on not missing affected patients. Overall, priorities for this guideline were informed by an international survey of stakeholders, including health care providers, patients, and caregivers, facilitated by the partner organizations.28
Methods
The guideline panel developed and graded the recommendations and assessed the certainty in the supporting evidence following the GRADE approach.4-9,29,30 The overall guideline-development process, including funding of the work, panel formation, management of conflicts of interest, internal and external review, and organizational approval, was guided by American Society of Hematology (ASH) policies and procedures derived from the G-I-N–McMaster Guideline Development Checklist (http://cebgrade.mcmaster.ca/guidecheck.html)31 and was intended to meet recommendations for trustworthy guidelines by the Institute of Medicine (IOM) and G-I-N.1-3
Organization, panel composition, planning, and coordination
These guidelines were developed as a collaboration by ASH, the International Society of Thrombosis and Haemostasis (ISTH), the National Hemophilia Foundation (NHF), and the World Federation of Hemophilia (WFH). The work of the panel was coordinated by ASH and the Outcomes and Implementation Research Unit at KUMC (funded by the collaborating organizations, under a paid agreement). Project oversight was provided by the ASH Guideline Oversight Subcommittee, which reported to the ASH Committee on Quality. All 4 collaborating organizations made nominations, with ASH vetting all individuals appointed to the guideline panel. The Outcomes and Implementation Research Unit at KUMC vetted and retained researchers to conduct systematic reviews of evidence and coordinate the guideline-development process including the use of the GRADE approach. The membership of the panel and the systematic review team is described in supplemental File 1.
The panel included pediatric and adult hematologists, internists, and laboratory specialists who all had clinical and research expertise on the guideline topic, as well as 4 patient representatives. One chair was a content expert; the other chair was an expert in guideline-development methodology. The panel also included a clinical vice-chair who served on both the diagnosis and management panels to ensure that their efforts were coordinated. All panelists were full and equal voting members with regard to the recommendations, with the exception of recusals as described in the next section.
In addition to synthesizing evidence systematically, the methods team from KUMC supported the guideline-development process, including determining methods, preparing meeting materials, and facilitating panel discussions. The panel’s work was done using Web-based tools (www.surveymonkey.com and www.gradepro.org) and face-to-face and online meetings.
Guideline funding and management of conflicts of interest
Development of these guidelines was wholly funded by the 4 collaborating organizations: ASH, ISTH, NHF, and WFH. Organization staff supported panel appointments and attended meetings but had no role in choosing the guideline questions or determining the recommendations.
Members of the guideline panel received travel reimbursement for attendance at in-person meetings. The patient representatives received an honorarium of $200 dollars each. Through the Outcomes and Implementation Research Unit at KUMC, some researchers who contributed to the systematic evidence reviews received salary or grant support. Other researchers participated to fulfill requirements of an academic degree or program.
Conflicts of interest of all participants were managed according to ASH policies based on IOM recommendations (2009) and G-I-N.3 Participants disclosed all financial and nonfinancial interests relevant to the guideline topic. ASH staff and the ASH Guideline Oversight Subcommittee reviewed the disclosures and composed the guideline panel to include a diversity of expertise and perspectives and avoid a majority of the panel having the same or similar conflicts. The greatest attention was given to direct financial conflicts with for-profit companies that could be directly affected by the guidelines. A majority of the guideline panel, including the cochairs, had no such conflicts. None of the researchers from the Outcomes and Implementation Research Unit at KUMC who contributed to the systematic evidence reviews or who supported the guideline-development process had any such conflicts.
Recusal was used to manage certain conflicts.5,32-34 During deliberations about recommendations, any panel member with a current direct financial conflict in a commercial entity that marketed any product that could be affected by a specific recommendation participated in discussions about the evidence and clinical context but was recused from making judgments or voting about individual domains (eg, magnitude of desirable consequences) and the direction and strength of the recommendation. The Evidence-to-Decision (EtD) framework for each recommendation describes which individuals were recused from making judgments about each recommendation.
In July 2020, 1 panelist disclosed that during the guideline-development process, he received a direct payment from a company that could be affected by the guidelines. The activity and disclosure occurred after the panel had agreed on recommendations; therefore, the panelist was not recused. Members of the Guideline Oversight Subcommittee reviewed the guidelines in relation to this late disclosure and agreed that conflict was unlikely to have influenced any of the recommendations.
Supplemental File 2 provides the complete disclosure-of-interest forms of all panel members. Individuals disclosed direct financial interests for 2 years prior to appointment in part A of the forms, indirect financial interests in part B, and relevant other interests (not mainly financial) in part C. Part D describes new interests disclosed by individuals after appointment. Part E summarizes ASH decisions about which interests were judged to be conflicts and how they were managed, including through recusal.
Supplemental File 3 provides the complete disclosure-of-interest forms of researchers who contributed to these guidelines.
Formulating specific clinical questions and determining outcomes of interest
The panel used the GRADEpro guideline-development tool (www.gradepro.org)35 and SurveyMonkey (www.surveymonkey.com) to brainstorm and then prioritize the questions described in Table 1. To generate the initial list of possible questions, a working group of clinicians, patients, and representatives from ASH, ISTH, NHF, and WFH was established prior to the formation of the guideline panel. A survey was developed by the KUMC methods team to prioritize these questions, and it was then translated from English into French and Spanish and widely publicized. Six hundred one participants responded from 71 countries, including clinicians, patients and caregivers, and members of allied health teams. Detailed information about this process, which determined the 10 questions to be included in the guideline, was published in 2019.28
The panel selected outcomes of interest for each question a priori, following the approach described in detail elsewhere.6 Although acknowledging considerable variation in the impact on patient outcomes, the panel considered the following outcomes as critical for clinical decision-making across questions: major bleeding, transfusion and treatment, gastrointestinal bleeding, blood loss, symptom severity, minor bleeding, mortality, and unnecessary testing. These outcomes will be affected by the accurate diagnosis of different subtypes of VWD and avoiding inaccurate mislabeling of patients.
Evidence review and development of recommendations
For each guideline question, the methods team from the Outcomes and Implementation Research Unit at KUMC prepared a GRADE EtD framework, using the GRADEpro guideline-development tool (www.gradepro.org).4,5,31 The EtD table summarized the results of systematic reviews of the literature that were updated or performed for this guideline. The EtD table addressed effects of interventions, resource utilization (cost-effectiveness), values and preferences (relative importance of outcomes), equity, acceptability, and feasibility. The guideline panel reviewed draft EtD tables before, during, or after the guideline panel meeting, made suggestions for corrections, and identified missing evidence. To ensure that recent studies were not missed, searches (presented in supplemental File 4) were updated on 8 January 2020, and panel members were asked to suggest any studies that might have been considered missed that fulfilled the inclusion criteria for the individual questions.
Under the direction of the Outcomes and Implementation Research Unit at KUMC, researchers followed the general methods outlined in the Cochrane Handbook for Systematic Reviews of Interventions (handbook.cochrane.org) for conducting updated or new systematic reviews of intervention effects. For new reviews, risk of bias was assessed at the health outcome level using the Cochrane Collaboration’s risk-of-bias tool for nonrandomized studies and the Quality Assessment of Diagnostic Accuracy Studies 2 (QUADAS-2) tool for test-accuracy studies.36 In addition to conducting systematic reviews of test accuracy, the researchers searched for evidence related to baseline risks, values, preferences, and costs, and summarized findings within the EtD frameworks.4,5,30 Subsequently, the certainty in the body of evidence (also known as quality of the evidence or confidence in the estimated effects) was assessed for test-accuracy outcomes following the GRADE approach based on the following domains: risk of bias; precision, consistency, and magnitude of the estimates of effects; directness of the evidence; and risk of publication bias. The certainty was categorized into 4 levels: very low (⊕◯◯◯), low (⊕⊕◯◯), moderate (⊕⊕⊕◯), and high (⊕⊕⊕⊕).7,8,29,30,37-39
During a 2-day in-person meeting followed by online communication and conference calls, the panel developed clinical recommendations based on the evidence summarized in the EtD tables. For each recommendation, the panel took a population perspective and came to consensus on the following: the certainty in the evidence, the balance of benefits and harms of the compared management options, and the assumptions about the values and preferences associated with the decision. The guideline panel also explicitly took into account the extent of resource use associated with alternative management options. The panel agreed on the recommendations (including direction and strength), remarks, and qualifications by consensus based on the balance of all desirable and undesirable consequences. The final guidelines, including recommendations, were reviewed and approved by all members of the panel.
Interpretation of strong and conditional recommendations
The recommendations are labeled as either “strong” or “conditional” according to the GRADE approach. The words “the guideline panel recommends” are used for strong recommendations, and “the guideline panel suggests” for conditional recommendations. Table 2 provides GRADE’s interpretation of strong and conditional recommendations by patients, clinicians, health care policy makers, and researchers.
Document review
Draft recommendations were reviewed by all members of the panel, revised, and then made available online on 6 April 2020 for external review by stakeholders including allied organizations, other medical professionals, patients, and the public; 51 individuals submitted comments. The document was revised to address pertinent comments, but no changes were made to recommendations. On 18 August 2020, the ASH Guideline Oversight Subcommittee confirmed that the defined guideline-development process had been followed; on 26 August 2020, the ASH Committee on Quality confirmed that the defined guideline-development process had been followed; and on 28 August 2020, the officers of the ASH Executive Committee approved submission of the guidelines for publication under the imprimatur of ASH. On 25 August 2020, the WFH confirmed that the defined guideline-development process had been followed; on 27 August 2020, the NHF confirmed that the defined guideline-development process had been followed; and on 28 August 2020, the ISTH confirmed that the defined guideline-development process had been followed. The guidelines were then subjected to peer review by Blood Advances.
How to use these guidelines
These guidelines are primarily intended to help clinicians make decisions about diagnostic and treatment alternatives. Other purposes are to inform policy, education, and advocacy, and to state future research needs. They may also be used by patients. These guidelines are not intended to serve or be construed as a standard of care. Clinicians must make decisions on the basis of the clinical presentation of each individual patient, ideally through a shared process that considers the patient’s values and preferences with respect to the anticipated outcomes of the chosen option. Decisions may be constrained by the realities of a specific clinical setting and local resources, including but not limited to institutional policies, time limitations, and availability of diagnostic tests and/or treatments. These guidelines may not include all appropriate methods of care for the clinical scenarios described. As science advances and new evidence becomes available, recommendations may become outdated. Following these guidelines cannot guarantee successful outcomes. ASH, ISTH, NHF, and WFH do not warrant or guarantee any products described in these guidelines.
Statements about the underlying values and preferences as well as qualifying remarks accompanying each recommendation are its integral parts and serve to facilitate more accurate interpretation. They should never be omitted when quoting or translating recommendations from these guidelines. Implementation of the guidelines will be facilitated by the related interactive forthcoming decision aids. The use of these guidelines is also facilitated by the links to the EtD frameworks and interactive summary-of-findings tables in each section.
Recommendations
Bleeding-assessment tools
For patients suspected of VWD, should a BAT or nonstandardized clinical assessment (not using a BAT) be used to screen for VWD?
For patients (especially men and children) suspected of VWD with a negative/normal bleeding score (based on a BAT), should blood testing be done or is no blood testing needed?
Recommendation 1
For patients with a low probability of VWD (eg, seen in the primary care setting), the panel recommends using a validated BAT as an initial screening test to determine who needs specific blood testing over nonstandardized clinical assessment (strong recommendation based on moderate certainty in the evidence from diagnostic accuracy studies ⊕⊕⊕◯).
Remarks:
This recommendation applies predominantly to adult women, as the data supporting the use of a BAT as a screening tool is strongest in this patient group.
The quality of nonstandardized clinical assessment will vary among the users of these guidelines.
Specific blood testing for VWD refers to VWF:Ag, platelet-dependent VWF activity (eg, VWF:GPIbM), and FVIII:C.
Recommendation 2
For patients with an intermediate probability of VWD (eg, referred to a hematologist), the panel suggests against relying on a BAT to decide whether to order specific blood testing (conditional recommendation based on moderate certainty in the evidence from diagnostic accuracy studies ⊕⊕⊕◯).
Remarks:
This recommendation addresses patients with an intermediate VWD pretest probability (∼20%) corresponding to those typically referred for hematology evaluation because of an abnormal personal bleeding history or abnormal initial laboratory tests (eg, prolonged aPTT) (including men and children).
Beyond their utility as a screening test in the primary care setting, BATs can be used in the referral setting to assess and document the severity of bleeding and can be used in conjunction with specific blood testing as part of the initial diagnostic approach.
Specific blood testing for VWD refers to VWF:Ag, platelet-dependent VWF activity (eg, VWF:GPIbM), and FVIII:C.
Recommendation 3
For patients with a high probability of VWD (eg, affected first-degree relative), the panel recommends against relying on a BAT to decide whether to order specific blood testing (strong recommendation based on moderate certainty in the evidence from diagnostic accuracy studies ⊕⊕⊕◯).
Remarks:
This recommendation addresses patients with a high VWD pretest probability (∼50%) corresponding to those typically referred for hematology evaluation because of an affected first-degree relative regardless of their bleeding symptoms or initial laboratory tests (including men and children).
Beyond their utility as a screening test in the primary care setting, BATs can be used in the referral setting to assess and document the severity of bleeding and can be used in conjunction with specific blood testing as part of the initial diagnostic approach.
Specific blood tests for VWD refer to VWF:Ag, platelet-dependent VWF activity (eg, VWF:GPIbM), and FVIII:C.
Summary of the evidence.
These recommendations were stratified on 3 VWD prevalences to account for the frequency of affected patients in different populations or clinical settings. Recommendation 1 is intended for primary care practitioners assuming a relatively low VWD prevalence in their clinic population, based on literature that shows an ∼3% prevalence of bleeding disorders in a population with abnormal laboratory tests (eg, prolonged aPTT).40 Recommendation 2 assumes a 20% VWD prevalence and is based on studies of consecutive patients referred to a hematology clinic, typically because of a personal history of bleeding/bruising and/or because of abnormal initial laboratory tests (eg, prolonged aPTT).41 Recommendation 3 assumes a 50% prevalence and is based on individuals with an affected first-degree relative, in keeping with the autosomal-dominant inheritance of most subtypes of the disease, regardless of bleeding symptoms or the results of initial laboratory tests.42,43 For these recommendations, 7 cohort studies that included 112 patients with a pooled sensitivity of 0.75 (95% confidence interval [CI], 0.66-0.83) were assessed and judged to be highly accurate; the sensitivity data were strongest for adult women. For specificity, the 7 cohort studies included 863 patients and had a pooled specificity of 0.54 (95% CI, 0.29-0.77) with moderate test accuracy.44-50 It is important to note that the included studies assessed the use of validated BATs vs not using a BAT rather than nonstandardized testing. A detailed review of validated BATs can be found at https://elearning.wfh.org/resource/compendium-of-assessment-tools/#bleeding_assessment_tools1a42-60ce78a1-2573f205-9a34. The EtD frameworks for these recommendations are available online at https://guidelines.ash.gradepro.org/profile/RBzFDJwKapc and https://guidelines.ash.gradepro.org/profile/aVdJ7pZVxu4.
Benefits, harms, and burden.
The primary benefit of a BAT is to identify patients who have VWD but would be missed without the use of this tool. Additionally, BATs provide a standardized approach to assessment. The panel considered not missing an affected patient an important benefit, in addition to identifying patients in a timely manner and decreasing unnecessary blood testing. Furthermore, BATs provide educational value to patients and clinical experts about bleeding symptoms and possible interventions, and offer validation to patients by recognizing symptoms of the disease. It is important to acknowledge that the identified studies using BATs as a screening tool included mostly women: they are most effective in this patient group.
The potential harms that could be caused by the use of a BAT include the possibility of missing affected individuals who have not manifested bleeding symptoms, such as men and children. This key issue is why, in higher-prevalence settings, BATs should be used in conjunction with specific blood testing rather than as the sole screening tool to decide whether blood testing should be done. BATs may also identify individuals who have bleeding disorders other than VWD; however, appropriate laboratory testing should distinguish those individuals. Additionally, some treatments will also be helpful for individuals with other bleeding disorders (such as tranexamic acid or combined oral contraceptives for heavy menstrual bleeding).
Other EtD criteria and considerations.
Published BATs are all freely available, but the administration of an expert-administered version takes time in the clinic (including the requirement of appropriate training and education for the individual administering the tool). The self-administered BAT (Self-BAT) addresses this issue; however, it is currently available only in English and French.46 The ISTH-BAT (which is expert-administered) has been translated and is available in German, Italian, Norwegian, Spanish,51 and Japanese (http://square.umin.ac.jp/kintenka/index.html). BATs are generally accepted by patients, as there is widespread familiarity with the completion of health-related questionnaires across many clinical settings. BATs (particularly expert-administered versions) may be less feasible in the primary care setting because of time and resource constraints.
Conclusions and research needs for these recommendations.
The panel determined that there is a moderate certainty of evidence for the test accuracy of the validated BATs. The panel agreed that there is a net benefit from the use of BATs in a low-prevalence setting but that in higher-prevalence settings, BATs should not be used as a sole screening test to determine who needs additional testing. Based on the available evidence, it is likely that the use of BATs will identify patients with VWD in primary care settings to help clinicians identify who needs additional, specialized laboratory testing. In other clinical settings, the use of BATs provides a standardized method for the documentation and assessment of the severity of bleeding symptoms as an adjunct to laboratory testing. With these recommendations, the panel considered published data from the use of a number of different BATs; however, many have evolved from the Vicenza bleeding questionnaire published in 2005 and have a high degree of overlap in their questions and the scoring systems.52 Additionally, the ISTH recently published a consensus BAT endorsed by that organization.53 The panel identified the need for future studies focused on the sensitivity and specificity of different score thresholds in the pediatric population (particularly the adolescent population) and in men.
Assays of platelet-dependent activity of VWF
For patients suspected of VWD, should the VWF:RCo assay (automated and nonautomated) or newer assays that reflect the platelet-binding activity of VWF (eg, VWF:GPIbM, VWF:GPIbR) be used to diagnose VWD?
Recommendation 4
The panel suggests newer assays that measure the platelet-binding activity of VWF (eg, VWF:GPIbM, VWF:GPIbR) over the VWF:RCo assay (automated or nonautomated) for the diagnosis of VWD (conditional recommendation based on low certainty in the evidence from diagnostic accuracy studies ⊕⊕◯◯).
Good practice statement: VWF activity assays should be performed in a laboratory with appropriate expertise.
Summary of the evidence.
A total of 13 studies were identified as relevant for this question; however, 6 informed the final recommendation. Data were reviewed for all published methods for VWF:RCo, VWF:GPIbM, VWF:GPIbR, and VWF:Ab (supplemental File 5); however, consistent with the recommendation of the ISTH and other groups, we focused our deliberations on the first 3 as direct measures of the platelet-binding activity of VWF.54,55 The ranges of sensitivity and specificity across 4 studies for VWF:RCo were 0.83 to 1.00 and 0.87 to 0.95, respectively.56-59 For VWF:GPIbR, from 4 studies, it was 0.80 to 1.00 and 0.81 to 0.9756-59 and for VWF:GPIbM, from 2 studies, 0.62 to 0.82 and 0.90 to 0.97, respectively.56-59 Therefore, the panel judged test accuracy to be generally comparable between the different assays. There was a serious risk of bias in all studies because of the case-control design, and only 2 studies reported on all 3 assays.56-59 Additionally, the published studies indirectly addressed the question because the tests were used to classify patients as opposed to making a new VWD diagnosis. The EtD framework for this recommendation is available online at https://guidelines.ash.gradepro.org/profile/VRjivq3oyEY.
Benefits, harms, and burden.
The panel judged there to be moderate benefits of the newer assays, reflecting the lower coefficient of variation and higher reproducibility compared with VWF:RCo. Additionally, although the published studies were comparable in terms of test accuracy, they did not include a large number of patients of African descent and therefore do not clearly reflect the presence of VWF variants in that population that can affect ristocetin binding with the VWF:RCo but do not affect VWF function or represent the true risk of bleeding (eg, the D1472H sequence variant).60,61 This creates the risk of overdiagnosis of these patients with VWF:RCo, which was considered a potential harm.
Other EtD criteria and considerations.
Although estimates vary between countries, in general, the price is comparable between assays but payer systems and levels of insurability vary widely. The specialized technical nature of these assays (and the required expertise to perform and interpret them) is another limitation to widespread availability, as are issues of US Food and Drug Administration (FDA) approval of the newer assays in the United States. Lastly, not all tests are available in all laboratories, and decisions to switch from one assay to another must take into account local circumstances.
Conclusions and research needs for this recommendation.
The guideline panel determined that there is low-certainty evidence for a net health benefit from using newer assays that measure the platelet-binding activity of VWF (eg, VWF:GPIbM, VWF:GPIbR) over VWF:RCo for patients suspected of having VWD. Other EtD criteria were generally in favor of using the newer assays. The panel identified the need for additional research focused on the performance of the assays in different racial/ethnic groups. Additionally, the panel identified the need for international guidance on detailed characteristics of appropriate laboratory expertise for VWF assays.
VWF levels that normalize with age
For patients with a historic diagnosis of type 1 VWD but who now have normal VWF levels, should the diagnosis of VWD be reconsidered, or should it be removed?
Recommendation 5
The panel suggests reconsidering the diagnosis as opposed to removing the diagnosis for patients with previously confirmed type 1 VWD who now have VWF levels that have normalized with age (conditional recommendation based on very low certainty in the evidence of effects ⊕◯◯◯).
Remarks:
With this recommendation, the panel worked under the assumption that the original diagnosis of type 1 VWD was accurate.
Aging and comorbidities are known to increase VWF levels. However, the association between the increased VWF levels and bleeding symptoms is not established.
Decisions about reconsidering or removing the diagnosis should consider the patient’s values and preferences and be informed by a shared decision-making process.
Summary of the evidence.
We identified 6 observational studies that indirectly addressed this question and show that ∼43% of VWD patients have VWF levels that normalize with age62-67 ; however, only 1 study adjusted for comorbidities that also could increase VWF levels.62 Additionally, no studies longitudinally evaluated whether the bleeding phenotype improved or resolved with increased VWF levels. The widespread lack of age-specific normal ranges was also identified as a complicating factor. The possibility that changes in the ability of laboratories to measure VWF:RCo could be playing a role was considered; however, in 1987, Gill et al published a cross-sectional study of blood donors that showed a 0.01 IU/mL annual increase in VWF levels between 20 and 60 years of age, suggesting that the levels truly do increase with age.68 Whether that is in relation to comorbidities or independent of them remains unclear. The EtD framework for this recommendation is available online at https://guidelines.ash.gradepro.org/profile/JHcpxNiXNGU.
Benefits, harms, and burden.
Reconsidering (as opposed to removing) the diagnosis would allow clinicians to consider and test for a concomitant bleeding disorder (eg, a platelet function disorder), particularly if this testing was not done at the time of the type 1 VWD diagnosis. The panel acknowledges that the degree of VWF normalization may influence management decisions for future bleeds/procedures and that clinicians may choose to use tranexamic acid alone and avoid desmopressin because of the concern of cardiovascular complications and/or thrombosis in older patients. The panel was concerned that a decision to remove a VWD diagnosis might result in a patient not receiving appropriate treatment of a bleed or prior to a procedure, in addition to the patient not having appropriate clinical follow-up and monitoring.
Other EtD criteria and considerations.
No specific resources would be required to remove a VWD diagnosis; however, the panel acknowledges that the necessary discussion between physician and patient is likely to be complicated and require adequate time. Additionally, removal of a diagnosis could have significant effects on insurance coverage in some countries. Reconsidering the diagnosis also requires a detailed discussion and may not completely avoid the issue of loss of insurance coverage; for example, in the United States, patients with a diagnosis of bleeding of unknown cause (BUC) are generally restricted from coverage of intranasal desmopressin. The panel recognized that patients may have widely different views on the acceptability of removing a VWD diagnosis. Patients with minimal bleeding symptoms are likely to be less concerned than those with significant bleeding, and a change in diagnosis may be less acceptable to the latter group. Regardless, clear communication and shared decision-making in this critical aspect of patient management is key, and one must keep in mind the physical and psychosocial impact on the patient.
Conclusions and research needs for this recommendation.
The panel determined that there is very low certainty evidence for a net health benefit by reconsidering as opposed to removing a VWD diagnosis for patients with previously confirmed VWD who now have VWF levels that have normalized with age. Multiple factors make a firm diagnosis of VWD difficult. It must be acknowledged that mildly reduced VWF:Ag and VWF:RCo or VWF:GPIbM levels do not always firmly establish a diagnosis of VWD; conversely, levels at the lower end of the normal range do not always exclude the diagnosis. Although the VWF:Ag assay has good precision and reproducibility, VWF:RCo has greater variability, resulting in the potential for misdiagnosis and/or misclassification. Data showing that an increase in VWF levels with age is accompanied by a decrease in bleeding risk/symptoms are not available, therefore, the removal of a VWD diagnosis is very difficult. The panel identified a critical need for longitudinal studies that correlate VWF levels with bleeding symptoms as patients age, adjusted for comorbidities.
Type 1 VWD
For patients with an abnormal initial VWD screen (low VWF:Ag and/or platelet-dependent VWF activity) suspected of type 1 VWD, should the diagnostic cutoff be at VWF:Ag and/or VWF platelet-dependent activity <0.30 IU/mL or <0.50 IU/mL?
Recommendation 6
The panel recommends a VWF level of <0.30 IU/mL regardless of bleeding, and for patients with abnormal bleeding, a VWF level of <0.50 IU/mL to confirm the diagnosis of type 1 VWD (strong recommendation based on low certainty in the evidence of effects ⊕⊕◯◯).
Remarks:
VWF level(s) refers to VWF:Ag and/or platelet-dependent VWF activity (eg, VWF:GPIbM).
The lower limit of the normal range as determined by the local laboratory should be used if it is <0.50 IU/mL. ABO-specific reference ranges are not required.
VWF is an acute-phase reactant that increases in response to a variety of stimuli (eg, bleed, trauma, pregnancy). VWD diagnostic testing should be performed when patients are at a baseline state of health.
Summary of the evidence.
A total of 9 observational studies were reviewed that address this question, including studies that evaluated the genetic basis of type 1 VWD,42,65,69,70 determined likelihood ratios (LRs) for type 1 VWD,42,71-73 correlated VWF levels with bleeding,65,71 and evaluated patients referred for investigation of a possible bleeding disorder.18,74 The cutoff value of 0.30 IU/mL was evaluated based on expert consensus and previous guideline recommendations.75 Patients with VWF levels <0.30 IU/mL were shown to have VWF mutations detected 75% to 82% of the time.69,70 In contrast, patients with VWF levels of 0.30 to 0.50 IU/mL had VWF mutations detected 44% to 60% of the time.65,69,70 In terms of LRs, patients with VWF levels of 0.30 to 0.40 IU/mL had an LR of infinity, as VWF was confirmed in all cases with second-level testing. For VWF levels of 0.41 to 0.50 IU/mL, the LR was 0.73 (0.41-1.30); for VWF levels of 0.51 to 0.60 IU/mL, the LR was 0.33 (0.18-0.62).71 Of critical importance, studies evaluating the correlation of VWF levels and bleeding symptoms show a similar bleeding phenotype across the range of VWF levels and specifically do not show more severe bleeding in those with VWF levels <0.30 IU/mL.65,69 Additionally, 70 of 93 patients with VWF levels of 0.30 to 0.50 IU/mL were investigated after a bleeding episode: mucocutaneous bleeding was present in 35, 25 bled after surgery, and 10 bled after dental procedures. Ten experienced >1 bleeding symptom.71 The EtD framework for this recommendation is available online at https://guidelines.ash.gradepro.org/profile/Ckc7oThe8q0.
Benefits, harms, and burdens.
The issue of diagnostic cutoffs is of great importance in certain health care systems, as it has a major impact on who is able to access care. The panel judged that those with VWF levels between 0.30 and 0.50 IU/mL and bleeding symptoms would have a net health benefit from a clear type 1 VWD diagnosis. The panel considered recommending that patients with VWF levels between 0.30 and 0.50 IU/mL and a positive family history should also be diagnosed with type 1 VWD (regardless of bleeding symptoms); however, the risk of an inaccurate diagnosis in a family member was felt to be a significant concern. Additionally, it was recognized that family structures vary widely and might influence the possibility of a diagnosis, for example, if someone had many female relatives who have heavy menstrual bleeding. Furthermore, although the inheritance of type 1 VWD in families with VWF levels <0.30 IU/mL is autosomal dominant, in families with VWF levels of 0.30 to 0.50 IU/mL, the issues of incomplete penetrance and variable expressivity complicate inheritance.70 In this latter group, the bleeding is likely to be complex, with contribution from genes outside of VWF; a concomitant bleeding disorder, such as a platelet function disorder, should be considered. These issues led to debate about the cause of bleeding in someone with milder reductions in VWF; however, these patients are still likely to benefit from the many treatments that are given for VWD, although they may suffer harms from side effects.
Other EtD criteria and considerations.
Diagnosing individuals with type 1 VWD who have VWF levels between 0.30 and 0.50 IU/mL might result in more repeat testing for those on the borderline. This could have accessibility and feasibility implications, particularly for those who do not live in centers with specialized coagulation laboratories.
Conclusions and research needs for this recommendation.
The panel determined that there is low-certainty evidence for a net health benefit of less restrictive diagnostic criteria for the diagnosis of type 1 VWD. Despite the low certainty in the evidence, the panel decided on a strong recommendation for 2 reasons: (1) a high value was placed on an explicit diagnosis to ensure access to care for those with a bleeding phenotype and (2) to ensure international uniformity in diagnostic criteria and the avoidance of center-specific thresholds based on a conditional recommendation.76 Although a definite diagnosis of type 1 VWD is straightforward in those with VWF levels <0.30 IU/mL, the advantage of pursuing and assigning a definitive diagnosis in mild or borderline cases was weighed against the risk of overdiagnosis and overmedicalization. As noted, the panel placed a high priority on not missing the diagnosis, especially for patients with bleeding symptoms, in order to ensure that management to treat/prevent bleeds is provided. Research priorities were identified including detailed data (including outcomes for bleeding with procedures and prevalence of a concomitant bleeding disorder) for patients with VWF levels between 0.30 and 0.60 IU/mL as well as the correlation with bleeding symptoms and information about family members of patients with type 1 VWD.
Type 1C VWD
For patients suspected of type 1 VWD with increased VWF clearance (type 1C VWD), should the ratio of propeptide to VWF antigen (VWFpp/VWF:Ag) or a desmopressin trial with 1- and 4-hour postinfusion blood work be used to confirm increased VWF clearance?
Recommendation 7
The panel suggests against using the VWFpp/VWF:Ag (ratio of VWF propeptide to antigen) and rather using a desmopressin trial with 1- and 4-hour postinfusion blood work to confirm increased VWF clearance for patients with VWD suspected of type 1C should be used (conditional recommendation based on low certainty in the evidence from diagnostic accuracy studies ⊕⊕◯◯).
Summary of the evidence.
A decrease in VWF survival (or increased VWF clearance) has long been suggested as a potential mechanism for type 1 VWD and is now known to account for ∼15% to 20% of cases.20,21 This was first clearly described for patients with Vicenza-type VWD, known to be caused by the VWF missense mutation R1205H.77 It was subsequently shown that this phenotype could be identified by an increased VWFpp/VWF:Ag ratio, given that the propeptide is stored in the Weibel-Palade bodies of endothelial cells in a 1:1 ratio with the mature protein but dissociates after secretion.78 If mature VWF is subject to enhanced clearance, the ratio of VWFpp to VWF:Ag increases. The response to desmopressin can also identify these patients, if a 4-hour postinfusion time point is included and shows a >30% decrease from the peak VWF level.79 Five studies were reviewed to address this question, including 2 that evaluated the correlation between the VWFpp/VWF:Ag ratio and VWF half-life78,79 and 3 that reported on the correlation of the VWFpp/VWF:Ag ratio and VWF mutation.21,22,80 No test-accuracy results were presented in the included studies because of the lack of an accepted reference standard to define type 1C VWD. In general, a higher VWFpp/VWF:Ag ratio was associated with a shorter VWF half-life and a higher rate of an identified VWF mutation, but it was noted that in some patients, the ratio can be normal but the clearance of VWF rapid.79 The EtD framework for this recommendation is available online at https://guidelines.ash.gradepro.org/profile/wBmLq8BFekg.
Benefits, harms, and burden.
The accurate identification of patients who have type 1C VWD has management implications, as these patients may require VWF concentrate to treat/prevent bleeds. A desmopressin trial, in addition to identifying patients with increased VWF clearance, also provides very useful information to the clinician about the utility of that treatment of an individual patient. Not all patients can safely undergo a desmopressin trial, including very young and very old patients, because of the risks associated with desmopressin (eg, hyponatremia or thrombosis); in these patients, the VWFpp/VWF:Ag ratio could be helpful.
Other EtD criteria and considerations.
Desmopressin trials require significant resources to complete in terms of nursing time, clinic space, and laboratory costs. Repeated blood draws are required, which could decrease acceptability for some patients. A significant time commitment is also required to complete the trial, including 1- and 4-hour postinfusion levels, resulting in patients needing to make an extra visit to clinic and miss a day of work or school; however, with a clear explanation of why the trial (and the 4-hour postinfusion level) is needed, it is likely that most patients would agree to have it performed. Lastly, protocols for the completion of desmopressin trials and definitions of responsiveness vary widely. Conversely, the VWFpp assay is simple to perform and requires only a single blood draw, but it is not available in most clinical laboratories.
Conclusions and research needs for this recommendation.
Overall, the panel determined that there is low certainty for a net health benefit from the use of a desmopressin trial over the VWFpp/VWF:Ag ratio for the identification of type 1C VWD. Feasibility and acceptability concerns for desmopressin trials were judged to be substantial, but this did not outweigh the lack of availability of the VWFpp assay. Research priorities include the need for studies addressing the sensitivity and specificity of various VWFpp/VWF:Ag thresholds, the clearance and half-life of VWFpp, and whether those variables are constant, in addition to studies that always include the 4-hour postinfusion time point for desmopressin trials.
Type 2 VWD
For patients with an abnormal initial VWD screen (low VWF:Ag and/or platelet-dependent VWF activity) suspected of type 2 VWD, should a platelet-dependent VWF activity/VWF:Ag ratio cutoff of <0.5 or a higher cutoff of <0.7 be used to confirm type 2 VWD?
Recommendation 8
The panel suggests against a platelet-dependent VWF activity/VWF:Ag ratio cutoff of <0.5 and rather using a higher cutoff of <0.7 should be used to confirm type 2 VWD (2A, 2B, or 2M) for patients with an abnormal initial VWD screen (conditional recommendation based on very low certainty in the evidence from diagnostic accuracy studies ⊕◯◯◯).
Remark:
Some patients with type 2 VWD have normal VWF:Ag and platelet-dependent VWF activity but a low ratio of platelet-dependent VWF activity/VWF:Ag.
Summary of the evidence.
Six observational studies were identified that addressed this question; all 6 evaluated diagnostic test accuracy,59,81-85 and 1 also looked at VWF mutations felt to be causative of type 2M VWD.83 The pooled sensitivity for the higher cutoff of <0.70 was 0.90 (95% CI, 0.83-0.94) compared with 0.58 to 0.79 for <0.5. Specificity was assumed to be 100% for <0.5 but was not directly available from the published studies. The panel judged there to be a serious risk of bias in 4 studies due to the case-control study design and serious unexplained heterogeneity between the studies. The EtD framework for this recommendation is available online at https://guidelines.ash.gradepro.org/profile/Orzbo0O_gbw.
Benefits, harms, and burden.
The panel was less concerned about false positives for this question, as additional testing, such as VWF multimer analysis, the VWF:CB assay, and/or genotyping, is typically performed for type 2 VWD, providing further clarification of the subtype. False negatives were judged to be of greater importance by both clinical and patient experts because of the concern of missing patients who would benefit from treatment. The false-negative rate was much higher for the <0.5 cutoff than the <0.7 cutoff (65 more false negatives of 1000 in a population with 30% prevalence). However, it should be noted that this question applies to patients with abnormal initial VWD testing, making it more likely that a patient would be misclassified as having type 1 VWD than missed altogether.
Other EtD criteria and considerations.
There would be no direct change to cost of using different ratio cutoffs for type 2 VWD; however, a higher cutoff is likely to result in more repeat testing and additional assays being performed. Lack of widespread availability and differing levels of insurance coverage of VWF assays were highlighted as issues, in addition to variability in terms of clinical and laboratory expertise about type 2 VWD. Problems with the VWF:RCo and false-positive results in the African American population were also noted.
Conclusions and research needs for this recommendation.
The guideline panel determined that there is low-certainty evidence for a net health benefit from using a VWF:RCo/VWF:Ag cutoff of <0.7 over a lower cutoff of <0.5 for patients suspected of type 2 VWD. Additional research is needed to understand variability in the VWF:RCo in different ethnic groups.
For patients suspected of type 2A, 2B, or 2M VWD in need of additional testing, should a VWF multimer analysis or a VWF:CB to VWF:Ag ratio (VWF:CB/VWF:Ag) be used?
Recommendation 9
The panel suggests either VWF multimer analysis or VWF:CB/VWF:Ag (ratio of VWF collagen binding to antigen) to diagnose type 2 VWD for patients suspected of type 2A, 2B, or 2M in need of additional testing (conditional recommendation based on very low certainty in the evidence from diagnostic studies ⊕◯◯◯).
Remark:
Most laboratories that do the VWF:CB assay use type I and/or III collagen, which is known to be a surrogate for the presence of high-molecular-weight VWF.
Summary of the evidence.
There were a total of 10 studies that addressed this question. Ten addressed diagnostic test accuracy for types 2A and 2B59,85-93 and 6 for type 2M.87-90,92,93 For types 2A and 2B, the sensitivity of VWF:CB/VWF:Ag was 0.90 (95% CI, 0.78-0.96) and specificity was 0.95 (95% CI, 0.89-0.98), vs a sensitivity of 0.90 (95% CI, 0.90-0.99) and specificity of 0.97 (95% CI, 0.94-0.99) for multimers. For type 2M VWD, the sensitivity of VWF:CB/VWF:Ag was 0.98 (95% CI, 0.96-1.00) with a specificity of 0.99 (95% CI, 0.98-1.00), vs a sensitivity of 0.86 (95% CI, 0.73-0.98) and a specificity of 0.97 (95% CI, 0.94-0.99) for multimers. However, there was a serious risk of bias in many of the studies because of the case-control design, and it must be acknowledged that the centers doing the multimer analysis for these studies were referral centers with significant experience and expertise. Additionally, there was no consistent cutoff for the VWF:CB/VWF:Ag ratio. Lastly, type 2M VWD is defined by a normal multimer profile, making this assay the reference standard for that subtype. The EtD framework for this recommendation is available online at https://guidelines.ash.gradepro.org/profile/26d0oeZn088.
Benefits, harms, and burden.
The accurate identification of patients with types 2A, 2B, or 2M is important for prognosis and for family counseling. Patients with type 2 VWD generally have more significant bleeding than those with type 1 VWD, and the family histories are clearly autosomal dominant, without the issues of incomplete penetrance or variable expressivity that complicate milder forms of type 1 VWD.45 Desmopressin is relatively contraindicated in type 2B VWD, as it may cause thrombocytopenia due to increased platelet binding; however, patients suspected of having this subtype would usually have additional testing performed, such as genetic testing, that would clarify the subtype.94 Patients with type 2A or 2M are less likely to respond to desmopressin, but this would be identified at the time of a desmopressin trial.95 Other treatment decisions are not likely to vary significantly between subtypes.
Other EtD criteria and considerations.
The VWF:CB assay is not widely available, and there are differences in the type of collagen used. Collagen types I and III interact with the A3 domain of VWF and type IV collagen with the A1 domain; as noted, most laboratories that perform this assay use type I and/or III collagen. VWF multimer analysis is technically challenging and is generally much more expensive than the VWF:CB assay. Insurance coverage for both assays varies widely.
Conclusions and research needs for this recommendation.
The panel determined that there is low-certainty evidence to recommend either performing VWF multimer analysis or using the VWF:CB/VWF:Ag ratio to identify type 2A, 2B, or 2M VWD. Additional research is needed to assess the diagnostic test accuracy of multimers in VWD patients who have known abnormal VWF:CB.
For patients suspected of type 2A or 2B VWD, should low-dose RIPA or targeted genetic testing be used to diagnose type 2B VWD?
Recommendation 10
The panel suggests targeted genetic testing over low-dose RIPA to diagnose type 2B VWD for patients suspected of type 2A or 2B in need of additional testing (Figure 2) (conditional recommendation based on low certainty in the evidence from diagnostic accuracy studies ⊕⊕◯◯).
Summary of the evidence.
We identified 15 studies that addressed this question, 14 that reported the identification of type 2B VWD mutations,84,94-106 and 9 that reported phenotype-genotype correlations in 2B VWD.84,94,97,98,100-103,105 The pooled sensitivities were 1.00 (95% CI, 1.00-1.00) for genetic testing and 0.99 (95% CI, 0.60-1.00) for RIPA; specificity was not available from the included studies. There was serious risk of bias because of the case-control study design and serious issues with the reference standard and/or index test bias in many of the studies; type 2B VWD is often defined by the VWF mutation and/or the identification of platelet agglutination with a low ristocetin concentration on RIPA. The EtD framework for this recommendation is available online at https://guidelines.ash.gradepro.org/profile/hGn1YO1dxh4.
Benefits, harms, and burden.
The accurate identification of type 2B VWD is important because it has relevance for prognosis and also treatment. Patients with this subtype typically have a more severe bleeding phenotype compared with other type 2 and type 1 VWD patients, and the bleeding risk has been shown to correlate with the degree of thrombocytopenia.94 Additionally, desmopressin is relatively contraindicated because it can worsen the thrombocytopenia.
Other EtD criteria and considerations.
Pathogenic VWF variants that cause type 2B VWD are found in exon 28; therefore, targeted genetic testing for well-characterized missense mutations is possible. Genetic testing is not available at all centers, but it is relatively straightforward to ship samples for this type of analysis. Likewise, RIPA is not available at all centers; however, that test requires a fresh sample; therefore, shipping is not possible, limiting accessibility to the test. Additionally, the methodology to perform a RIPA is not standardized, and different concentrations of ristocetin have been proposed as indicating a positive test (eg, 0.5 mg/mL and 0.25 mg/mL). In general, patients accept having either test performed; however, there are some patient groups that have concerns about genetic tests (eg, some First Nations people). Genetic testing is typically more expensive than RIPA, and insurance coverage is variable.
Conclusions and research needs.
The panel determined that there is low-certainty evidence for a net health benefit from the use of targeted genetic testing (when available) over RIPA to diagnose type 2B VWD. Additional research, focused on the diagnostic test accuracy of RIPA, would be beneficial.
For patients suspected of type 2N VWD in need of additional testing, should VWF:FVIIIB or targeted genetic testing be used to diagnose type 2N VWD?
Recommendation 11
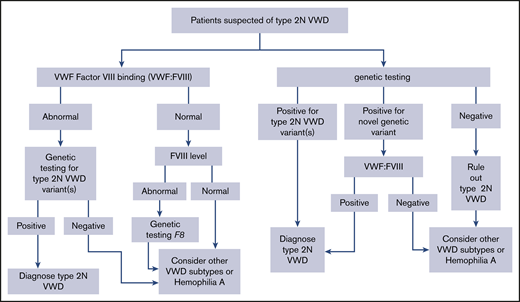
The panel suggests using either VWF:FVIIIB or targeted genetic testing (when available) for patients with suspected type 2N VWD in need of additional testing (Figure 3) (conditional recommendation based on low certainty in the evidence from diagnostic accuracy studies ⊕⊕◯◯).
Summary of the evidence.
We identified 17 studies that addressed this question, 16 that reported on the identification of a type 2N mutation57,103,105,107-119 and 13 that reported the correlation between assays.57,102,103,105,107,109-111,114-117,119 The sensitivities of both genetic testing and the VWF:FVIIIB assay were reported as 1.00 (95% CI, 1.00-1.00); however, both were used as the reference standard, resulting in serious bias. Specificity was not available from the included studies. There was also serious patient-selection bias due to the case-control study design in all studies. The EtD framework for this recommendation is available online at https://guidelines.ash.gradepro.org/profile/HDVamZn5f-0.
Benefits, harms, and burden.
The accurate identification of type 2N VWD is important for a number of reasons. In contrast to all other type 2 and type 1 VWD, type 2N is autosomal recessive, which is critical for appropriate genetic counseling within families. Type 2N VWD can be mistaken for hemophilia A because of the low FVIII level, with critical treatment implications: patients with hemophilia A are treated with FVIII concentrate; however, type 2N VWD patients require VWF replacement therapy to prevent/treat serious bleeds. Pathogenic VWF variants that cause type 2N VWD are generally found in exons 18 to 20; however, novel variants that have not been previously described or characterized can be found.120 In those cases, evidence of the phenotype is required (decreased binding of FVIII by VWF, which would be identified by the VWF:FVIIIB assay). However, doing only the VWF:FVIIIB assay will not be as informative as genetic testing for family counseling, as patients with type 2N VWD can be homozygous for a single 2N variant or compound heterozygous for 2 2N variants (and in both cases a child would be heterozygous for a 2N allele and unlikely to manifest bleeding); sometimes only a single 2N variant is found, in which case it is assumed that there is coinheritance of a null allele (therefore, a child could be heterozygous for 2N with no bleeding or could have type 1 VWD). Additionally, if an F8 variant is identified as opposed to a VWF variant, then the diagnosis can be confirmed as hemophilia A with appropriate treatment and genetic counseling for X-linked inheritance.120
Other EtD criteria and considerations.
Genetic testing is not available at all centers, but it is relatively straightforward to ship samples to reference laboratories for this type of analysis. Likewise, VWF:FVIIIB is not available at all centers, but shipping of plasma is possible. In general, patients accept having either test performed; however, there are some patient groups that have concerns about genetic testing (eg, some First Nations people). Genetic testing is more expensive than the VWF:FVIIIB assay, and insurance coverage is variable.
Conclusions and research needs for this recommendation.
The panel determined that there is low-certainty evidence to suggest using either VWF:FVIIIB or genetic testing for patients with suspected type 2N VWD. Indeed, the panel agreed that the tests can be complementary in the diagnostic workup of patients. Identifying a reference standard for type 2N VWD was identified as a research priority.
What are others saying and what is new in these guidelines?
Multiple published guidelines recommend a cutoff of <0.30 IU/mL for a definite diagnosis of type 1 VWD, with the US National Institutes of Health National Heart, Lung, and Blood Institute (NHLBI) guidelines stating that “this recommendation does not preclude the diagnosis of VWD in individuals with VWF:RCo of 30 to 50 IU/dL if there is supporting clinical evidence and/or family evidence for VWD.”75 The recommendation of the United Kingdom Haemophilia Centre Doctors Organization (UKHCDO) states: “Patients with an appropriate bleeding history and VWF activity 0.30-0.50 IU/mL should be regarded as having primary hemostatic bleeding with reduced VWF as a risk factor rather than VWD. We suggest referring to this as ‘Low VWF’.”55 This guideline panel prioritized ensuring access to medical care and therefore recommends a level of <0.30 IU/mL regardless of bleeding and a VWF level of 0.30 to 0.50 IU/mL for patients with abnormal bleeding to confirm the diagnosis of type 1 VWD. There was significant concern that the term “Low VWF” creates an unintentional barrier to patients receiving appropriate care because of a lack of a clear diagnosis, particularly in countries without universal health care systems. A patient’s bleeding symptoms were the primary consideration for this recommendation.
Recently, the NHF Medical and Scientific Advisory Council (MASAC) raised concern about the critical importance of preanalytical variables in VWD testing, and the possibility of false-positive results from samples processed or shipped improperly.121 This guideline panel acknowledges the significance of this issue in avoiding a misdiagnosis of VWD, which has been reported in a cohort of women being investigated for a bleeding disorder.122
These guidelines focused on the most common inherited forms of VWD; however, we would like to highlight the recently published guidance document on the diagnosis and management of platelet-type VWD from the ISTH Platelet Physiology Subcommittee123 and several recent reviews on acquired von Willebrand syndrome.124,125
Limitations of these guidelines
The limitations of these guidelines are inherent in the low or very low certainty in the evidence identified for many of the questions.
Plans for updating these guidelines
After publication of these guidelines, the collaborating organizations will maintain them through surveillance for new evidence, ongoing review by experts, and regular revisions based on literature searches.
Updating or adapting recommendations locally
Adaptation of these guidelines will be necessary in many circumstances. These adaptations should be based on the associated EtD frameworks.10
Data for the Evidence-to-Decision frameworks will be publicly available via Web links from the online version of the document.
Acknowledgments
The authors acknowledge:
William L. Nichols Jr, who served as chair of the NHLBI von Willebrand Disease Expert Panel and of the 2007 publication “The Diagnosis, Evaluation, and Management of von Willebrand Disease,” for advice and guidance during the early stages of this guideline’s development.
Jenny Castano, Emily Senerth, and Rob Kunkle from ASH; Cary Clark from ISTH; Ellen Riker and Mark Skinner from NHF; and Fiona Robinson from WFH for organizational support.
The ASH ISTH NHF WFH von Willebrand Disease Guideline Scoping Group:
Paula James, Chair
Nathan T. Connell, Vice-chair
Jeroen Eikenboom, ISTH representative
Magdy El Ekiaby, WFH representative
Veronica Flood, NHF representative
Jessica Graham, NHF patient representative
Susan Halimeh, WFH patient representative
Barbara Konkle, NHF representative
Frank Leebeek, ISTH representative
William L. Nichols Jr, ASH representative
Flora Peyvandi, WFH representative
Francesca Stufano for participating in guideline question prioritization and outcome selection.
Omar Abughanimeh, Abdalla El Alayli, Osama Diab, Ahmad Dimassi, Bader Madoukh, Aref Qureini, and Sammy Tayiem for their work on the systematic review team.
These guidelines are dedicated to the memory of J. Evan Sadler, a member of the VWD management guideline panel who died in December 2018. His work challenged the community to think critically about the diagnosis of VWD. The panel is grateful for his advice and contributions to the early guideline efforts, and for his lifetime of scholarship and service to the bleeding disorders community.
Authorship
Contribution: P.D.J. wrote the first draft of the manuscript and revised the manuscript based on authors’ suggestions; guideline panel members B.A., J.D.P., J.E., N.G., S.H., V.J.-P., B.K., C.M., S.M., R.R.M., J.S.O., N.S., and R.S. critically reviewed the manuscript and provided suggestions for improvement; knowledge synthesis team members M.A.K., N.H., and R.A.M. contributed evidence summaries to the guidelines, critically reviewed the manuscript, and provided suggestions for improvement; P.D.J., N.T.C., and R.A.M. were the cochairs of the panel and led panel meetings; V.H.F. chaired the concurrent guideline panel on management of VWD, critically reviewed the manuscript, and provided suggestions for improvement; and all authors approved the content.
Conflict-of-interest disclosure: Authors were members of the diagnosis guideline panel or members of the systematic review team or both, with the addition of V.H.F. who chaired the management panel. All completed disclosure-of-interest forms, which were reviewed by ASH and are available as supplemental Files 2 and 3.
Correspondence: Paula D. James, Queen’s University, Room 2015 Etherington Hall, 94 Stuart St, Kingston, ON K7L 3N6, Canada; e-mail: jamesp@queensu.ca.