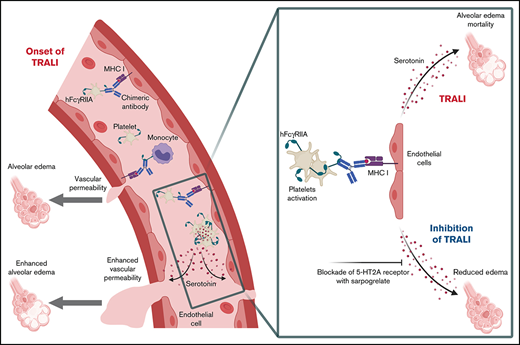
Key Points
Platelet FcγRIIA/CD32A activation exacerbates the severity of antibody-mediated TRALI.
Platelet serotonin release and activation of 5-HT2A serotonin receptors is responsible for the FcγRIIA-dependent exacerbation of TRALI.

Abstract
Transfusion-related acute lung injury (TRALI) remains a major cause of transfusion-related fatalities. The mechanism of human antibody-mediated TRALI, especially the involvement of the Fcγ receptors, is not clearly established. Contrary to mice, human platelets are unique in their expression of the FcγRIIA/CD32A receptor, suggesting that our understanding of the pathogenesis of antibody-mediated TRALI is partial, as the current murine models incompletely recapitulate the human immunology. We evaluated the role of FcγRIIA/CD32A in TRALI using a humanized mouse model expressing the FcγRIIA/CD32A receptor. When challenged with a recombinant chimeric human immunoglobulin G1/mouse anti–major histocompatibility complex class I monoclonal antibody, these mice exhibited exacerbated alveolar edema and higher mortality compared with wild-type (WT) mice. Unlike in WT mice, monocytes/macrophages in CD32A+ mice were accessory for TRALI initiation, indicating the decisive contribution of another cell type. Platelet activation was dramatically increased in CD32A+ animals, resulting in their increased consumption and massive release of their granule contents. Platelet depletion prevented the exacerbation of TRALI in CD32A+ mice but did not affect TRALI in WT animals. By blocking platelet serotonin uptake with fluoxetine, we showed that the severity of TRALI in CD32A+ mice resulted from the serotonin released by the activated platelets. Furthermore, inhibition of 5-hydroxytryptamine 2A serotonin receptor with sarpogrelate, before or after the induction of TRALI, abolished the aggravation of lung edema in CD32A+ mice. Our findings show that platelet FcγRIIA/CD32A activation exacerbates antibody-mediated TRALI and provide a rationale for designing prophylactic and therapeutic strategies targeting the serotonin pathway to attenuate TRALI in patients.
Introduction
Transfusion-related acute lung injury (TRALI) is a syndrome characterized by the development of non-cardiogenic pulmonary edema. TRALI contributes significantly to the morbidity of transfusion and can even be fatal.1-3 TRALI can result from the presence in the transfused product of alloantibodies specific for polymorphic HLA/major histocompatibility complex class I (MHC I) or MHC class II molecules or for neutrophil antigens, the latter 2 constituting the major causes of mortality in this syndrome. The in situ detection of platelets and neutrophils in the lungs of patients who died of TRALI has led to the proposition that these cells participate in its pathogenesis.4
To analyze the mechanisms of alloantibody-mediated TRALI, several models have been developed in H-2d BALB/c mice, all of which use the anti–H-2Dd/H-2Kd MHC I mouse monoclonal antibody (mAb) 34-1-2S. In a two-hit model, an inflammatory priming step, induced by intraperitoneal administration of a low dose of lipopolysaccharides (LPS; 0.1 mg/kg), is followed 24 hours later by intravenous injection of 34-1-2S (0.5 mg/kg).5 In one-hit models, the animals are not sensitized with LPS, but at least 10-fold higher amounts of the antibody need to be injected to induce TRALI.6,7 These models have allowed assessment of the respective contributions of the different blood cell populations and have resulted in various conclusions. Monocytes/macrophages were found to be essential, whereas CD4+ T cells and dendritic cells behaved as negative regulators of TRALI; depending on the studies, neutrophils and platelets were described as being either required or dispensable for lung edema formation.5-15 Recently, endothelial MHC I has been identified as the critical molecular target for initiation of anti–MHC I mAb-mediated lung injury.16
One challenge with respect to the mechanisms of antibody-mediated TRALI is to elucidate the biological features of the interactions between the Fc part of the immunoglobulin G (IgG) subclasses and their specific Fcγ receptors (FcγRs) present on immune cells. In different studies, FcγRs were found to be major9,13,17 or accessory7 mediators of lung injury induced by anti–MHC I. Concerning the repertoire of FcγRs, one major difference between man and mouse is the presence in the human genome of the FCGR2A gene, encoding an activating low-affinity receptor for IgG subclasses called FcγRIIA, also known as CD32A.18 This receptor is expressed on various immune cells of myeloid origin susceptible to participation in antibody-mediated TRALI responses, namely neutrophils, monocytes, macrophages, dendritic cells, and platelets. It bears an intracellular immunoreceptor tyrosine-based activation motif that mediates cell activation upon its crosslinking by IgG-immune complexes (IgG-ICs).19 Unlike leukocytes, which express various types of FcγR, human platelets possess only FcγRIIA/CD32A.
It is well established that IgG-ICs can strongly activate human platelets, resulting in their aggregation and the release of platelet granule contents.20-22 In contrast, FcγRIIA/CD32A is absent from mice and mouse platelets, devoid of any FcγR, cannot be activated by IgG-ICs. Thus, in the current murine models of antibody-mediated TRALI, FcγR-dependent responses would appear to be biased toward the involvement of circulating leukocytes and/or macrophages, while ignoring the potential contribution of FcγR-mediated platelet responses to the pathogenesis of TRALI in man. Accordingly, we have reported that in WT animals, FcγR+ patrolling monocytes and/or macrophages are required, whereas FcγR– platelets are dispensable for the early steps of experimental antibody-induced TRALI leading to the formation of lung edema.8,12 These concerns prompted us to make use of transgenic mice expressing human FcγRIIA/CD32A, especially as these mice have been shown to reliably mimic several human physiological and pathological responses. Indeed, in these animals, platelet FcγRIIA/CD32A participates not only in protective immune responses20 but also in severe IgG-IC–dependent anaphylaxis,21,23 heparin-induced thrombocytopenia,24 and sepsis.25
The current study explored the role of FcγRIIA/CD32A in the pathogenesis of TRALI by using a new model of antibody-mediated TRALI induced by LPS sensitization and challenge with a recombinant chimeric human/mouse anti-mouse MHC I (H-2Dd/H-2Kd) mAb in mice expressing a human FcγRIIA/CD32A transgene. This strategy revealed a crucial contribution of platelet FcγRIIA/CD32A to antibody-induced TRALI through platelet activation and serotonin release, with exacerbation of TRALI responses, thereby opening up a new dimension in our understanding of the pathogenesis of TRALI in humans.
Methods
Mice
Transgenic mice expressing human FcγRIIA/CD32A (C57BL/6, H-2b)26 were kindly provided by P. Bruhns (Institut Pasteur) and bred in our animal facilities. These animals were crossed with BALB/cByJ mice (H-2d) (JAX, Charles River Laboratories). The presence of the hCD32A transgene was assessed by polymerase chain reaction analysis of genomic DNA using the primers 5'-CAATTTTGCTGCTATGGGC-3' and 5'-CTGGTCAAGGTCACATTCTTC-3'. TRALI experiments were performed on the F1 progeny (C57BL/6 x BALB/c) using 6- to 7-week-old male mice expressing the human FcγRIIA/CD32A transgene (CD32A+) and their WT littermates (CD32A–). Mice were housed in the animal facilities of the Etablissement Français du Sang–Grand Est (agreement N°E67-482-10). Experiments were approved by the Regional Committee for Ethics in Animal Experimentation of Strasbourg (CREMEAS, CEEA-35) and authorized by the French Ministry of Research, according to the regulations of the European Community (APAFIS#2018100815412773).
Two-event TRALI model
A previously described two-hit model5 was used, with a few modifications. Male mice were primed with an intraperitoneal injection of LPS (0.1 mg/kg body weight) and 24 hours later, anesthetized with xylazine (20 mg/kg body weight) and ketamine (100 mg/kg body weight). TRALI was induced by retro-orbital injection of the recombinant chimeric human IgG1 (hIgG1)/mouse 34-1-2S anti–MHC I mAb (anti–H-2Dd/H-2Kd, 1.5 mg/kg). The animals were maintained on warming plate heated to 37°C and euthanized 10 to 15 minutes or 2 hours after injection of the mAb. TRALI responses were assessed by measuring the protein content of bronchoalveolar lavages (BALs) in right lungs and by analyzing the pulmonary lesions in left lungs by histochemistry.8 Animals used as negative controls received saline. To visualize vascular leakage, 1% Evans Blue (50 µL) was injected intravenously before injection of the anti–MHC I mAb. All experiments were performed by operators blind to the genotype of the mice.
Statistical analysis
Statistical analyses were performed with GraphPad software (GraphPad Prism 5.02). Survival was analyzed by using the log-rank test. All other values were reported as the mean ± standard error of the mean and analyzed by using one- or two-way analysis of variance followed by a Dunn’s test or a Bonferroni post hoc analysis, depending on the normal distribution of the data established by using the Shapiro-Wilk test. For Figures 3A-B, 4, and 5A, E, and G, statistical analyses were performed without considering the negative controls (ie, mice injected with saline instead of the anti–MHC I mAb) because the n values of the negative controls were lower than those of the other conditions. These low n values of the negative control animals could be justified by the high reproducibility of the measurements between independent experiments and the ethical requirement to reduce the number of animals.
An extended methods section is available in the supplemental Materials.
Results
FcγRIIA/CD32A expression severely aggravates alveolar edema and increases mortality of antibody-mediated TRALI
To set up a humanized model of TRALI allowing evaluation of the biological activity of CD32A, we first cloned the variable regions of the heavy and light chains of the mouse mAb 34-1-2S and then fused them to the constant regions of the hIgG1 heavy chain and κ light chain, respectively, thus generating a chimeric human/mouse anti–MHC I mAb, called hIgG1-34-1-2S. hIgG1 was preferred to other IgG subclasses on account of its biological activities, which better resemble those of mouse IgG2a, together with its higher affinity for CD32A compared with other hIgG subclasses.27 Indeed, both the recombinant mAb hIgG1-34-1-2S and the original mouse mAb IgG2a-34-1-2S bound to H-2Kd MHC I with comparable affinity and stained circulating CD45+ leukocytes from BALB/c mice with the same efficacy (supplemental Figure 1A-B). In addition, hIgG1-34-1-2S– and mIgG2a-34-1-2S–containing immune complexes readily bound to FcγRIIA/CD32A and all mouse FcγRs-expressing CHO transfectants, whereas monomeric hIgG1-34-1-2S or mIgG2a-34-1-2S only bound efficiently to mFcγRI- and FcγRIV-expressing cells (supplemental Figure 1C). Therefore, we may anticipate that hIgG1-34-1-2S can activate all endogenous mouse FcγRs, in addition to FcγRIIA/CD32A.
hIgG1-34-1-2S was administered to LPS-sensitized (0.1 mg/kg, intraperitoneal) CD32A– and CD32A+ mice,5 and lung alveolar edema, a typical feature of experimental TRALI, was evaluated by quantifying the proteins in BALs 15 minutes later. The protein content of BALs increased significantly in CD32A– and CD32A+ mice compared with negative control animals. The increase was greater in CD32A+ mice, reaching twice the levels in CD32A– animals (CD32A+ vs CD32A–, 1.11 ± 0.14 mg/mL vs 0.48 ± 0.09 mg/mL, respectively; P < .01), indicating that the alveolar edema was more severe in CD32A+ mice than in CD32A– mice (Figure 1A). Histologic evaluation indicated that acute lung injury was also significantly increased in CD32A+ mice compared with CD32A– mice (Figure 1B). The enhanced pulmonary vascular permeability in CD32A+ animals was confirmed by measurement of the extravasation of Evans Blue dye into the lung tissues and BALs 15 minutes after injection of hIgG1-34-1-2S (Figure 1C). This increased vascular permeability seemed to be specific to the lungs, as no extravasation of the dye could be observed in other vascular territories of CD32A+ mice such as the glabrous skin of the ears, snout, or paws (Figure 1D). Within 2 hours following hIgG1-34-1-2S injection, 40% of the CD32A– mice died; this percentage increased to 90% in CD32A+ mice (log-rank test, P = .0027) (Figure 1E, left panel). At death, or at the 2-hour time point, alveolar edema was also more severe in CD32A+ mice than in CD32A– mice (Figure 1E, right panel). Altogether, these results indicate that FcγRIIA/CD32A expression exacerbates the disruption of the pulmonary endothelial barrier provoked by antibody-mediated TRALI, leading to reduced survival.
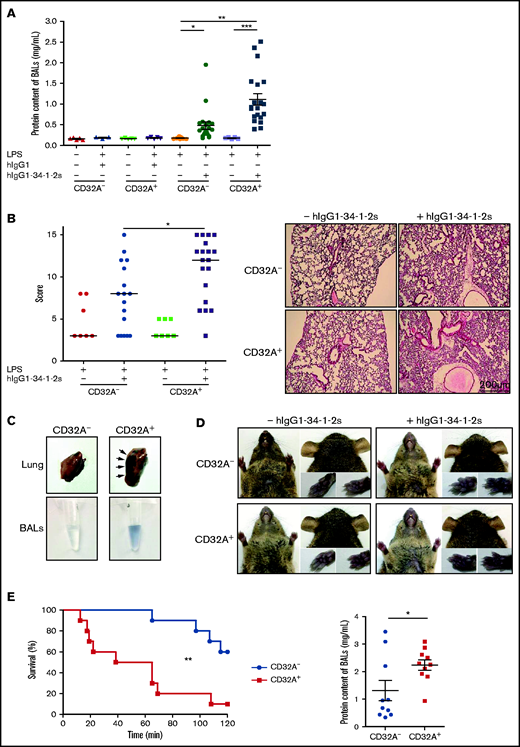
FcγRIIA/CD32A expression severely aggravates the alveolar edema and increases the mortality of antibody-mediated TRALI. The pathogenesis of TRALI was analyzed 15 minutes or 2 hours after injection of the chimeric human/mouse anti–MHC I mAb hIgG1-34-1-2S (1.5 mg/kg, intravenous) into LPS-sensitized (0.1 mg/kg, intraperitoneal) CD32A– or CD32A+ mice. (A) Protein concentrations in BALs were measured 15 minutes after injection of hIgG1-34-1-2S or in negative controls (CD32A–, n = 9-20; CD32A+, n = 3-20). (B) The histologic features of lung injury were determined 15 minutes after injection of hIgG1-34-1-2S or in negative controls (CD32A–, n = 7-17; CD32A+, n = 7-19). Scale bar = 200 µm. (C) Exemplary photographs showing increased pulmonary vascular permeability in CD32A+ mice compared with CD32A– mice revealed by extravasation of Evans Blue dye into the lung tissue and BALs 15 minutes after administration of hIgG1-34-1-2S. (D) Extravasation of Evans Blue dye was not observed in the vascular territories of the glabrous skin of the ears, snout, or paws 15 minutes after injection of hIgG1-34-1-2S or saline into CD32A+ or CD32A– mice. (E) Left: Kaplan-Meier survival plots for CD32A– and CD32A+ mice (n = 10) (log-rank test, P = .0027). Right: Protein concentrations in BALs of CD32A– and CD32A+ mice at death or at the 2-hour end point. Results are presented as the mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001 by one-way analysis of variance (panels A and B) or a Student t test (panel E, right).
FcγRIIA/CD32A expression severely aggravates the alveolar edema and increases the mortality of antibody-mediated TRALI. The pathogenesis of TRALI was analyzed 15 minutes or 2 hours after injection of the chimeric human/mouse anti–MHC I mAb hIgG1-34-1-2S (1.5 mg/kg, intravenous) into LPS-sensitized (0.1 mg/kg, intraperitoneal) CD32A– or CD32A+ mice. (A) Protein concentrations in BALs were measured 15 minutes after injection of hIgG1-34-1-2S or in negative controls (CD32A–, n = 9-20; CD32A+, n = 3-20). (B) The histologic features of lung injury were determined 15 minutes after injection of hIgG1-34-1-2S or in negative controls (CD32A–, n = 7-17; CD32A+, n = 7-19). Scale bar = 200 µm. (C) Exemplary photographs showing increased pulmonary vascular permeability in CD32A+ mice compared with CD32A– mice revealed by extravasation of Evans Blue dye into the lung tissue and BALs 15 minutes after administration of hIgG1-34-1-2S. (D) Extravasation of Evans Blue dye was not observed in the vascular territories of the glabrous skin of the ears, snout, or paws 15 minutes after injection of hIgG1-34-1-2S or saline into CD32A+ or CD32A– mice. (E) Left: Kaplan-Meier survival plots for CD32A– and CD32A+ mice (n = 10) (log-rank test, P = .0027). Right: Protein concentrations in BALs of CD32A– and CD32A+ mice at death or at the 2-hour end point. Results are presented as the mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001 by one-way analysis of variance (panels A and B) or a Student t test (panel E, right).
FcγRIIA/CD32A expression increases the activation of circulating neutrophils and platelets during antibody-mediated TRALI
Because all blood cell populations expressing CD32A could potentially contribute to this effect, we first evaluated the activation state of circulating leukocytes and platelets during TRALI. Baseline blood cell counts were similar between CD32A– and CD32A+ mice (supplemental Table 1). Within 15 minutes of injection of hIgG1-34-1-2S, plasma levels of the granular protein myeloperoxidase were significantly higher in CD32A+ mice compared with CD32A– mice (Figure 2A), suggesting an increased activation of blood neutrophils in CD32A+ mice. Immunohistology using an anti-mouse neutrophil antibody showed increased neutrophil sequestration in the lungs of CD32A+ mice compared with CD32A– mice (Figure 2B). In addition, the drop in circulating platelet count was more severe in CD32A+ mice than in CD32A– mice (Figure 2C), suggesting enhanced platelet activation in CD32A+ animals. Indeed, the circulating platelets of CD32A+ mice exhibited higher CD62P expression compared with those of CD32A– mice (Figure 2D). Immunohistology using an anti-mouse platelet antibody provided evidence for increased platelet sequestration in the lungs of CD32A+ animals compared with CD32A– animals (Figure 2E). Histology also revealed the presence of a few occlusive thrombi in the pulmonary microcirculation but in similar proportions in CD32A+ and CD32A– mice (supplemental Figure 2), ruling out a major contribution of thrombi to the increased edema formation. Thus, the activation of platelets and neutrophils was quantitatively more important in CD32A+ animals experiencing antibody-mediated TRALI.
![FcγRIIA/CD32A expression increases the activation of circulating neutrophils and platelets during antibody-mediated TRALI. LPS-sensitized (0.1 mg/kg, intraperitoneal) CD32A– or CD32A+ mice were injected with the mAb hIgG1-34-1-2S (1.5 mg/kg, intravenous) or vehicle and examined 15 minutes later. (A) Blood plasma levels of myeloperoxidase (MPO) (CD32A–, n = 5-11; CD32A+, n = 5-12). (B) Left: Neutrophil areas in the lungs of CD32A– and CD32A+ mice (n = 3). Right: Representative lung sections from CD32A– and CD32A+ mice immunolabeled with the anti-neutrophil mAb 1A8 (scale bar = 50 µm). (C) Circulating platelet counts before administration of LPS, 24 hours later, and 15 minutes after hIgG1-34-1-2S challenge (n = 8). (D) Percentage of CD62P-positive platelets (n = 6). (E) Left: Platelet areas in the lungs of CD32A– and CD32A+ mice (n = 3). Right: Representative lung sections from CD32A– and CD32A+ mice immunolabeled with the anti-platelet mAb RAM.1 (scale bar = 50 µm). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), *P < .05, **P < .01, ***P < .001 by one-way analysis of variance (panels A and D), two-way analysis of variance (panel C), or a Student t test (panels B and E).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/23/10.1182_bloodadvances.2021004336/2/m_advancesadv2021004336f2.png?Expires=1764214692&Signature=JRpOX3d8TYYZERB1QrMm3qXL8Hn0hGSAPgca55CxSgel55hcD595V0D~-UGadvWpX~3mQ5FNklCWtG4s-WUSonXCv6sARIoayePJM74la9SDzjNLNxY4CbcU9DGKJoCG34bxz-DPofDvc3agnztvDN~~LULperAskBxxEn6XOi0jb9Kp7bF5aDQP9OYngzlkybBkb23r0aOgUOA7HlMcR1s4IdeEyTwqDizCcYZC-UO7OqYv-ZN-4t4DYc0q-eakYKbkJ84yy-updKmpS1pUc7Khayx7O1GoXfP2-gEOjX7oZcLSsHiPYRX-6cFm-qeDqOxui3-Xp~5VyslRFValVw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
FcγRIIA/CD32A expression increases the activation of circulating neutrophils and platelets during antibody-mediated TRALI. LPS-sensitized (0.1 mg/kg, intraperitoneal) CD32A– or CD32A+ mice were injected with the mAb hIgG1-34-1-2S (1.5 mg/kg, intravenous) or vehicle and examined 15 minutes later. (A) Blood plasma levels of myeloperoxidase (MPO) (CD32A–, n = 5-11; CD32A+, n = 5-12). (B) Left: Neutrophil areas in the lungs of CD32A– and CD32A+ mice (n = 3). Right: Representative lung sections from CD32A– and CD32A+ mice immunolabeled with the anti-neutrophil mAb 1A8 (scale bar = 50 µm). (C) Circulating platelet counts before administration of LPS, 24 hours later, and 15 minutes after hIgG1-34-1-2S challenge (n = 8). (D) Percentage of CD62P-positive platelets (n = 6). (E) Left: Platelet areas in the lungs of CD32A– and CD32A+ mice (n = 3). Right: Representative lung sections from CD32A– and CD32A+ mice immunolabeled with the anti-platelet mAb RAM.1 (scale bar = 50 µm). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), *P < .05, **P < .01, ***P < .001 by one-way analysis of variance (panels A and D), two-way analysis of variance (panel C), or a Student t test (panels B and E).
FcγRIIA/CD32A expression increases the activation of circulating neutrophils and platelets during antibody-mediated TRALI. LPS-sensitized (0.1 mg/kg, intraperitoneal) CD32A– or CD32A+ mice were injected with the mAb hIgG1-34-1-2S (1.5 mg/kg, intravenous) or vehicle and examined 15 minutes later. (A) Blood plasma levels of myeloperoxidase (MPO) (CD32A–, n = 5-11; CD32A+, n = 5-12). (B) Left: Neutrophil areas in the lungs of CD32A– and CD32A+ mice (n = 3). Right: Representative lung sections from CD32A– and CD32A+ mice immunolabeled with the anti-neutrophil mAb 1A8 (scale bar = 50 µm). (C) Circulating platelet counts before administration of LPS, 24 hours later, and 15 minutes after hIgG1-34-1-2S challenge (n = 8). (D) Percentage of CD62P-positive platelets (n = 6). (E) Left: Platelet areas in the lungs of CD32A– and CD32A+ mice (n = 3). Right: Representative lung sections from CD32A– and CD32A+ mice immunolabeled with the anti-platelet mAb RAM.1 (scale bar = 50 µm). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), *P < .05, **P < .01, ***P < .001 by one-way analysis of variance (panels A and D), two-way analysis of variance (panel C), or a Student t test (panels B and E).
Monocytes/macrophages are required in WT mice but only accessory in CD32A+ mice for the initiation of antibody-mediated TRALI
Given the increase in the activation state and pulmonary sequestration of neutrophils in CD32A+ mice during TRALI, we looked at whether these cells contributed to the aggravation of lung injury. Neutrophil depletion (supplemental Figure 3A-B) had no significant impact on TRALI in CD32A– mice (Figure 3A), in agreement with our previous observations in the context of TRALI induced by the mIgG2a mAb 34-1-2S in WT mice.12 Depletion of neutrophils in CD32A+ animals did not reduce but rather slightly increased the protein content of BALs, an effect that cannot be easily explained. In any case, neutrophils did not seem to be responsible for the aggravation of alveolar edema in this humanized model of antibody-mediated TRALI.
![Monocytes/macrophages are required in WT mice but only accessory in CD32A+ mice for the initiation of antibody-mediated TRALI. (A) The Gr-1 mAb RB6-8C5 or control rIgG2bk (0.5 mg/kg, intravenous [IV]) was administered 24 hours before LPS (0.1 mg/kg, intraperitoneal) and protein concentrations in BALs were analyzed 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into CD32A+ (n = 3-16) or CD32A– (n = 3-16) mice. (B) Clodronate liposomes (2 mL/kg, IV), or vehicle, were injected 6 hours before hIgG1-34-1-2S challenge (1.5 mg/kg, IV) into LPS-sensitized (0.1 mg/kg, intraperitoneal) CD32A– or CD32A+ mice. Protein concentrations in BALs were evaluated 15 minutes later (CD32A–, n = 7-12; CD32A+, n = 4-13). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), *P < .05, ***P < .001 by one-way analysis of variance.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/23/10.1182_bloodadvances.2021004336/2/m_advancesadv2021004336f3.png?Expires=1764214692&Signature=Cx1~j2tmvNFzUFgJJQQbZlfZEjZUnr97jdIzOHdbZLFKcHhTk2G0WWziJKNvUNrGVDGn-5vxYu3oYaJA3z3kIHUec0I6Uf2p1CeXIBpOZqT2UhkOt6W8MxD-ONZOVB6kDrzRLA0sVNvnQ26tCAurGNgOrBwsfYY~O~8oayokXpg8NyVF4-Pb5rjfKuQXZu00Z5D9xQFCpg340olAf6zx-w3XF2nDCiB8GHFz81CSa43BYzn8VloGJbhLPi8rPPhR0P5HV1lx-3uQZrgNwH~PG42m8hdNYap3Yxgz2OIatoSHzHnh4WyPJpUymaJFnl-LdnuZekeGgQnk9KXuG6lsyw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Monocytes/macrophages are required in WT mice but only accessory in CD32A+ mice for the initiation of antibody-mediated TRALI. (A) The Gr-1 mAb RB6-8C5 or control rIgG2bk (0.5 mg/kg, intravenous [IV]) was administered 24 hours before LPS (0.1 mg/kg, intraperitoneal) and protein concentrations in BALs were analyzed 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into CD32A+ (n = 3-16) or CD32A– (n = 3-16) mice. (B) Clodronate liposomes (2 mL/kg, IV), or vehicle, were injected 6 hours before hIgG1-34-1-2S challenge (1.5 mg/kg, IV) into LPS-sensitized (0.1 mg/kg, intraperitoneal) CD32A– or CD32A+ mice. Protein concentrations in BALs were evaluated 15 minutes later (CD32A–, n = 7-12; CD32A+, n = 4-13). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), *P < .05, ***P < .001 by one-way analysis of variance.
Monocytes/macrophages are required in WT mice but only accessory in CD32A+ mice for the initiation of antibody-mediated TRALI. (A) The Gr-1 mAb RB6-8C5 or control rIgG2bk (0.5 mg/kg, intravenous [IV]) was administered 24 hours before LPS (0.1 mg/kg, intraperitoneal) and protein concentrations in BALs were analyzed 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into CD32A+ (n = 3-16) or CD32A– (n = 3-16) mice. (B) Clodronate liposomes (2 mL/kg, IV), or vehicle, were injected 6 hours before hIgG1-34-1-2S challenge (1.5 mg/kg, IV) into LPS-sensitized (0.1 mg/kg, intraperitoneal) CD32A– or CD32A+ mice. Protein concentrations in BALs were evaluated 15 minutes later (CD32A–, n = 7-12; CD32A+, n = 4-13). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), *P < .05, ***P < .001 by one-way analysis of variance.
We next investigated whether monocytes/macrophages contributed to lung injury in CD32A+ mice. These cells were eliminated by using clodronate liposomes, delivered 18 hours after LPS (ie, 6 hours before anti–MHC I mAb injection to allow inflammation to proceed). Under these conditions, TRALI was totally abolished in CD32A– mice (Figure 3B), in agreement with our previous observations in the context of TRALI induced by the mIgG2a mAb 34-1-2S in WT mice.12 In contrast, depletion of monocytes/macrophages in CD32A+ animals only partially reduced the protein content of BALs, indicating that these cells played an accessory role and thereby indicate the decisive intervention of another cell population.
Platelets are required for the FcγRIIA/CD32A–induced aggravation of antibody-mediated TRALI
The enhanced activation of platelets during TRALI in CD32A+ mice raised the question as to whether platelet activation or consumption was a marker of disease severity or actively contributed to the aggravation of TRALI. To explore the role of platelets in TRALI, we depleted circulating platelets using the anti-GPIbα mAb cocktail R300, 48 hours before inducing TRALI (supplemental Figure 4). Under these conditions, the alveolar edema associated with hIgG1-34-1-2S–mediated TRALI was not modified in CD32A– mice (Figure 4), consistent with our previously published work showing that platelets are dispensable for TRALI induced by the mIgG2a mAb 34-1-2S in WT mice.8 In contrast, the protein content of BALs was significantly reduced in thrombocytopenic CD32A+ mice, falling to the level observed in CD32A– animals, although remaining significantly above resting levels. These results showed that platelet FcγRIIA/CD32A activation exacerbates antibody-mediated TRALI.
![Platelets are required for the FcγRIIA/CD32A–induced aggravation of antibody-mediated TRALI. The mAb R300 or control rIgG2a (0.5 mg/kg, intravenous) was administered 24 hours before LPS (0.1 mg/kg, intraperitoneal) and protein concentrations in BALs were analyzed 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, intravenous) or vehicle into CD32A– or CD32A+ mice (n ≥ 9). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), *P < .05, **P < .01, ***P < .001 by one-way analysis of variance.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/23/10.1182_bloodadvances.2021004336/2/m_advancesadv2021004336f4.png?Expires=1764214692&Signature=N8zesLBYLoNfrp23gXYP8zkhNJV97Hgeb6iUvfa2s0oVUSt~BmYlg~x1YZ~CPcf5Wc4BAWcbPKdlINW-4zLLcGt2q8s9eNSiyWTGvpYEl0aHmIMHE7yROmfn2OneHXDnLdR5CJV8Rf1ohyXRKP5JbPQgKam8ltu3aZCVX73FBMyZrN1eQy51hM86dbCLoN72WkYeNrFIkgkFTNazLkaR875Ng~uL9YREFz-sH52Pu1kSvUXThUBuV2nEkUhGRlD-Iwq51~nKnm0Ls44-~TekeciHtkW2-T0JIpTydTtIndgxReYgBJxJVGo~S-MSHbvroYxBRGIqz0YxKXhXjo3IvA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Platelets are required for the FcγRIIA/CD32A–induced aggravation of antibody-mediated TRALI. The mAb R300 or control rIgG2a (0.5 mg/kg, intravenous) was administered 24 hours before LPS (0.1 mg/kg, intraperitoneal) and protein concentrations in BALs were analyzed 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, intravenous) or vehicle into CD32A– or CD32A+ mice (n ≥ 9). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), *P < .05, **P < .01, ***P < .001 by one-way analysis of variance.
Platelets are required for the FcγRIIA/CD32A–induced aggravation of antibody-mediated TRALI. The mAb R300 or control rIgG2a (0.5 mg/kg, intravenous) was administered 24 hours before LPS (0.1 mg/kg, intraperitoneal) and protein concentrations in BALs were analyzed 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, intravenous) or vehicle into CD32A– or CD32A+ mice (n ≥ 9). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), *P < .05, **P < .01, ***P < .001 by one-way analysis of variance.
Platelet-released serotonin, but not platelet-activating factor, is responsible for the FcγRIIA/CD32A–induced aggravation of antibody-mediated TRALI
We next wondered whether a mediator derived from platelets or released from their granules might be responsible for this effect. Platelet-activating factor, which is produced by activated platelets and plays a key role during anaphylaxis,28 was not involved in platelet FcγRIIA/CD32A–mediated aggravation of TRALI because the platelet-activating factor receptor antagonist ABT-491 had no impact on the protein content of BALs in either CD32A– or CD32A+ mice (Figure 5A).
![Platelet-released serotonin, but not platelet-activating factor, is responsible for the FcγRIIA/CD32A–induced aggravation of antibody-mediated TRALI. (A) The platelet-activating factor receptor antagonist ABT-491 (1.25 mg/kg, intravenous [IV]) or saline was administered 5 minutes before injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, intraperitoneal [IP]) CD32A– or CD32A+ mice, and protein concentrations in BALs were analyzed 15 minutes later (CD32A–, n = 3-8; CD32A+, n = 3-7). Plasma concentrations of platelet factor 4 (PF4) (CD32A–, n = 3-12; CD32A+, n = 5-12) (B) and serotonin (CD32A–, n = 3-12; CD32A+, n = 5-12) (C) 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice. (D) Serotonin concentrations in the serum of CD32A– or CD32A+ mice treated or not with the serotonin transporter inhibitor fluoxetine (160 mg/L) in drinking water for 3 weeks (CD32A–, n = 11-19; CD32A+, n = 5-18). (E) CD32A– or CD32A+ mice, treated or not with fluoxetine, were sensitized with LPS (0.1 mg/kg, IP) and, 24 hours later, injected with the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle. Protein concentrations in BALs were evaluated 15 minutes afterward (CD32A–, n = 3-16; CD32A+, n = 3-16). (F) Circulating platelet counts before and 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with fluoxetine (n ≥ 10). (G) Plasma concentrations of serotonin before and 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with clodronate (CD32A–, n = 4-6; CD32A+, n = 4-6). (H) Protein concentrations in BALs were evaluated 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with fluoxetine or clodronate (CD32A–, n = 5-9; CD32A+, n = 3-9). (I) Circulating platelet counts before and 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with clodronate (CD32A–, n = 7-12; CD32A+, n = 4-12). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), *P < .05, **P < .01, ***P < .001 by one-way analysis of variance.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/23/10.1182_bloodadvances.2021004336/2/m_advancesadv2021004336f5a.png?Expires=1764214692&Signature=ffw8syu6qvt2hufyhxUOzwuifYRO4aLWX07Gckogq8UB8HZbXz7iMCzzCw5rw42A13xzs4w9UHGkI107XYyMLVVSk~emVztPPi~JX7oMMm8LDtt4aSSLJ4wSyYAihXrcTRYgAqt-61T~Pas-4ksD-n97WpOaefIGcS8-KvNeYWGt~8NsisWC18o2AAw-4-4DcOQTuXgU5gJ13wOSWTThZvZ5TfJvv4FTYumGvspR-AUkIhtrjqkOxviukYC1moU4M4A3jkciyFiDxy1P5FxZPJYiiuDs2pPjxZG1fkJeJZt4wVi7DKsJuBQSGStG~m0vu2R~rbwv~9OfXyYhQ9JvcQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Platelet-released serotonin, but not platelet-activating factor, is responsible for the FcγRIIA/CD32A–induced aggravation of antibody-mediated TRALI. (A) The platelet-activating factor receptor antagonist ABT-491 (1.25 mg/kg, intravenous [IV]) or saline was administered 5 minutes before injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, intraperitoneal [IP]) CD32A– or CD32A+ mice, and protein concentrations in BALs were analyzed 15 minutes later (CD32A–, n = 3-8; CD32A+, n = 3-7). Plasma concentrations of platelet factor 4 (PF4) (CD32A–, n = 3-12; CD32A+, n = 5-12) (B) and serotonin (CD32A–, n = 3-12; CD32A+, n = 5-12) (C) 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice. (D) Serotonin concentrations in the serum of CD32A– or CD32A+ mice treated or not with the serotonin transporter inhibitor fluoxetine (160 mg/L) in drinking water for 3 weeks (CD32A–, n = 11-19; CD32A+, n = 5-18). (E) CD32A– or CD32A+ mice, treated or not with fluoxetine, were sensitized with LPS (0.1 mg/kg, IP) and, 24 hours later, injected with the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle. Protein concentrations in BALs were evaluated 15 minutes afterward (CD32A–, n = 3-16; CD32A+, n = 3-16). (F) Circulating platelet counts before and 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with fluoxetine (n ≥ 10). (G) Plasma concentrations of serotonin before and 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with clodronate (CD32A–, n = 4-6; CD32A+, n = 4-6). (H) Protein concentrations in BALs were evaluated 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with fluoxetine or clodronate (CD32A–, n = 5-9; CD32A+, n = 3-9). (I) Circulating platelet counts before and 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with clodronate (CD32A–, n = 7-12; CD32A+, n = 4-12). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), *P < .05, **P < .01, ***P < .001 by one-way analysis of variance.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/23/10.1182_bloodadvances.2021004336/2/m_advancesadv2021004336f5b.png?Expires=1764214692&Signature=1QzlWy9O4KyIsiUfDLaJWJ2iEeOrbGOKnGR~dRbMrsdFz4FSl~JoZdS4Bp6rlQ6OyW8W0bcWk9H9cxRWHordIp~DIn~kTOkz4Rd7gkggCpfkzlEAurO4wkNVFP-Nr~EUWdfNXETG9WmtROFOU2PUQ5Vh9pFALNTDdrGxIpC9KAn5PdvjHhFYnmMXuYICyGR1JaHHiSpF53Q0k4Q0ZCTy2jnm-HbcBer3Idcy7M~RuOEPkTxPhBW-rGD~pz6WdIff9aQ6qyacnJ~Xq-r6N7aWaTBXZl75gcPQXvjbGATEb9CKWkGJaeeXoeOPD-tzN4fDr0R1gpgsPuKBqF-VtdAkKw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Platelet-released serotonin, but not platelet-activating factor, is responsible for the FcγRIIA/CD32A–induced aggravation of antibody-mediated TRALI. (A) The platelet-activating factor receptor antagonist ABT-491 (1.25 mg/kg, intravenous [IV]) or saline was administered 5 minutes before injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, intraperitoneal [IP]) CD32A– or CD32A+ mice, and protein concentrations in BALs were analyzed 15 minutes later (CD32A–, n = 3-8; CD32A+, n = 3-7). Plasma concentrations of platelet factor 4 (PF4) (CD32A–, n = 3-12; CD32A+, n = 5-12) (B) and serotonin (CD32A–, n = 3-12; CD32A+, n = 5-12) (C) 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice. (D) Serotonin concentrations in the serum of CD32A– or CD32A+ mice treated or not with the serotonin transporter inhibitor fluoxetine (160 mg/L) in drinking water for 3 weeks (CD32A–, n = 11-19; CD32A+, n = 5-18). (E) CD32A– or CD32A+ mice, treated or not with fluoxetine, were sensitized with LPS (0.1 mg/kg, IP) and, 24 hours later, injected with the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle. Protein concentrations in BALs were evaluated 15 minutes afterward (CD32A–, n = 3-16; CD32A+, n = 3-16). (F) Circulating platelet counts before and 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with fluoxetine (n ≥ 10). (G) Plasma concentrations of serotonin before and 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with clodronate (CD32A–, n = 4-6; CD32A+, n = 4-6). (H) Protein concentrations in BALs were evaluated 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with fluoxetine or clodronate (CD32A–, n = 5-9; CD32A+, n = 3-9). (I) Circulating platelet counts before and 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with clodronate (CD32A–, n = 7-12; CD32A+, n = 4-12). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), *P < .05, **P < .01, ***P < .001 by one-way analysis of variance.
Platelet-released serotonin, but not platelet-activating factor, is responsible for the FcγRIIA/CD32A–induced aggravation of antibody-mediated TRALI. (A) The platelet-activating factor receptor antagonist ABT-491 (1.25 mg/kg, intravenous [IV]) or saline was administered 5 minutes before injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, intraperitoneal [IP]) CD32A– or CD32A+ mice, and protein concentrations in BALs were analyzed 15 minutes later (CD32A–, n = 3-8; CD32A+, n = 3-7). Plasma concentrations of platelet factor 4 (PF4) (CD32A–, n = 3-12; CD32A+, n = 5-12) (B) and serotonin (CD32A–, n = 3-12; CD32A+, n = 5-12) (C) 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice. (D) Serotonin concentrations in the serum of CD32A– or CD32A+ mice treated or not with the serotonin transporter inhibitor fluoxetine (160 mg/L) in drinking water for 3 weeks (CD32A–, n = 11-19; CD32A+, n = 5-18). (E) CD32A– or CD32A+ mice, treated or not with fluoxetine, were sensitized with LPS (0.1 mg/kg, IP) and, 24 hours later, injected with the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle. Protein concentrations in BALs were evaluated 15 minutes afterward (CD32A–, n = 3-16; CD32A+, n = 3-16). (F) Circulating platelet counts before and 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with fluoxetine (n ≥ 10). (G) Plasma concentrations of serotonin before and 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with clodronate (CD32A–, n = 4-6; CD32A+, n = 4-6). (H) Protein concentrations in BALs were evaluated 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with fluoxetine or clodronate (CD32A–, n = 5-9; CD32A+, n = 3-9). (I) Circulating platelet counts before and 15 minutes after injection of the mAb hIgG1-34-1-2S (1.5 mg/kg, IV) or vehicle into LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice treated or not with clodronate (CD32A–, n = 7-12; CD32A+, n = 4-12). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), *P < .05, **P < .01, ***P < .001 by one-way analysis of variance.
We next evaluated whether platelet granule contents were released during TRALI. Within 15 minutes of the onset of TRALI, platelet factor 4 (Figure 5B) and serotonin (Figure 5C), stored in alpha and dense granules, respectively, and released during activation-induced platelet degranulation, were detected at high concentrations in the plasma of CD32A+ animals but not of CD32A– animals. Serotonin, a known inducer of vascular permeability,29-31 has recently been identified by us and others as playing a critical part in platelet-dependent IgG-mediated anaphylaxis in CD32A+ transgenic animals.21,25 The vast majority of the total serotonin pool of the body is stored in platelet-dense granules.32 Platelets do not synthesize serotonin but use a serotonin transporter to capture plasma serotonin. Blocking platelet serotonin uptake with the serotonin transporter inhibitor fluoxetine resulted in almost complete depletion of the serotonin content of dense granules (Figure 5D). Whereas fluoxetine treatment had no observable impact on the lung edema in CD32A– mice, it abolished its aggravation in CD32A+ mice (Figure 5E). These findings indicate that platelet-released serotonin was responsible for the FcγRIIA/CD32A–dependent exacerbation of TRALI. One may note that the exacerbation of TRALI-associated thrombocytopenia still occurred in serotonin-depleted CD32A+ mice (Figure 5F), suggesting that serotonin, although a platelet activator, was not responsible for this effect.
Interestingly, release of platelet serotonin occurred normally during the residual TRALI in clodronate-treated CD32A+ mice (Figure 5G), suggesting that FcγRIIA/CD32A–mediated platelet activation and serotonin release was sufficient to initiate TRALI in transgenic animals. Conversely, monocyte/macrophage depletion completely abolished TRALI in fluoxetine-treated CD32A+ mice (Figure 5H), strongly suggesting that release of serotonin from platelets was responsible for the low- grade TRALI responses in clodronate-treated CD32A+ animals. The TRALI-associated thrombocytopenia remained exacerbated in CD32A+ mice with or without clodronate treatment (Figure 5I). In contrast, TRALI-associated platelet consumption was inhibited in CD32A– mice treated with clodronate, indicating that in WT animals, but not in transgenic ones, platelet consumption partly relied on monocytes and/or macrophages.
Blockade of the 5-hydroxytryptamine 2A serotonin receptor prevents the exacerbation of antibody-mediated TRALI in CD32A+ mice
Finally, we investigated how the serotonin released by platelets expressing FcγRIIA/CD32A could aggravate the development of TRALI-associated lung edema. Among the numerous serotonin receptor subtypes present on circulating blood cells and cells of the vessel wall, the 5-hydroxytryptamine (5-HT) 2A receptor has been shown to mediate the increase in vascular permeability occurring during IgG-IC–induced CD32A-mediated shock.25 To explore the involvement of this receptor in the serotonin-induced exacerbation of lung edema in CD32A+ mice, the selective 5-HT2A antagonist sarpogrelate was used.33 Intravenous injection of sarpogrelate 5 minutes before injection of hIgG1-34-1-2S had no significant effect on lung edema in CD32A– mice but prevented its aggravation in CD32A+ animals (Figure 6A). The 5-HT2A serotonin receptor therefore seemed to be responsible for the serotonin-induced exacerbation of FcγRIIA/CD32A–dependent TRALI.
![Blockade of the 5-HT2A serotonin receptor prevents the exacerbation of antibody-mediated TRALI in CD32A+ mice. (A) The selective 5-HT2A serotonin receptor antagonist sarpogrelate (1 mg/kg, intravenous [IV]) or saline was administered 5 minutes before hIgG1-34-1-2S challenge (1.5 mg/kg, IV) in LPS-sensitized (0.1 mg/kg, intraperitoneal [IP]) CD32A– or CD32A+ mice. Protein concentrations in BALs were evaluated 15 minutes later (n = 6). (B) Pulmonary elastance (Ers) in LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice after injection of the mAb hIgG1-34-1-2S (0.75 mg/kg, IV, n = 4) or vehicle (n = 3). (C) Sarpogrelate (1 mg/kg, IV) or saline was administered 5 minutes after hIgG1-34-1-2S challenge (1.5 mg/kg, IV) in LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice. Protein concentrations in BALs were evaluated 15 minutes later (n ≥ 22). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), **P < .01 by one-way analysis of variance (panels A and C).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/23/10.1182_bloodadvances.2021004336/2/m_advancesadv2021004336f6.png?Expires=1764214692&Signature=f3Uh2GLSUJF5cGTn2iFlNdDTNG8b9x4PtkPEM4T9Z2QpxNk-xOEAfeK3kDDQuSz8qknTWQX26f5l3kX8Edoaja67NGoJpKHnRpozGXa1tfyZBSuAyh97SqsaqZkV-sxYA0gCPUvP-ku4Rr6IDzKLADmVIOGKEXX21N28nP4TjY76I09u807oAnlhhuRzxZeE1Kc7txJvmoiBaPD1DUos8quu5gsYnripZhMM0kungIB2Ng-xv2Z9f3B75xeHF2TUmOum9zn1If4tKOVaRDg1cWQjDbnOaD2O-d3Q3tho6vJ0gwqkCZ-FgH2Byj-9ui1A~mvJZi9Ga~iLeJJR206n0Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Blockade of the 5-HT2A serotonin receptor prevents the exacerbation of antibody-mediated TRALI in CD32A+ mice. (A) The selective 5-HT2A serotonin receptor antagonist sarpogrelate (1 mg/kg, intravenous [IV]) or saline was administered 5 minutes before hIgG1-34-1-2S challenge (1.5 mg/kg, IV) in LPS-sensitized (0.1 mg/kg, intraperitoneal [IP]) CD32A– or CD32A+ mice. Protein concentrations in BALs were evaluated 15 minutes later (n = 6). (B) Pulmonary elastance (Ers) in LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice after injection of the mAb hIgG1-34-1-2S (0.75 mg/kg, IV, n = 4) or vehicle (n = 3). (C) Sarpogrelate (1 mg/kg, IV) or saline was administered 5 minutes after hIgG1-34-1-2S challenge (1.5 mg/kg, IV) in LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice. Protein concentrations in BALs were evaluated 15 minutes later (n ≥ 22). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), **P < .01 by one-way analysis of variance (panels A and C).
Blockade of the 5-HT2A serotonin receptor prevents the exacerbation of antibody-mediated TRALI in CD32A+ mice. (A) The selective 5-HT2A serotonin receptor antagonist sarpogrelate (1 mg/kg, intravenous [IV]) or saline was administered 5 minutes before hIgG1-34-1-2S challenge (1.5 mg/kg, IV) in LPS-sensitized (0.1 mg/kg, intraperitoneal [IP]) CD32A– or CD32A+ mice. Protein concentrations in BALs were evaluated 15 minutes later (n = 6). (B) Pulmonary elastance (Ers) in LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice after injection of the mAb hIgG1-34-1-2S (0.75 mg/kg, IV, n = 4) or vehicle (n = 3). (C) Sarpogrelate (1 mg/kg, IV) or saline was administered 5 minutes after hIgG1-34-1-2S challenge (1.5 mg/kg, IV) in LPS-sensitized (0.1 mg/kg, IP) CD32A– or CD32A+ mice. Protein concentrations in BALs were evaluated 15 minutes later (n ≥ 22). Results are presented as the mean ± standard error of the mean. P > .05 (not significant [ns]), **P < .01 by one-way analysis of variance (panels A and C).
These results suggest that the 5-HT2A receptor could represent a pharmacologic target to attenuate TRALI in humans. To test this hypothesis, we first checked the kinetics of TRALI development by measuring lung respiration parameters. Five minutes after challenge with hIgG1-34-1-2S, CD32A+ animals exhibited increased signs of respiratory distress, as evidenced by their enhanced pulmonary elastance compared with CD32A– mice (Figure 6B). Thus, when sarpogrelate was injected at this time point, it effectively abolished the aggravation of lung edema occurring in CD32A+ mice but not in CD32A– animals (Figure 6C). These findings showed that sarpogrelate prevents the exacerbation of TRALI mediated by platelet FcγRIIA/CD32A, which would provide a rationale for targeting serotonin receptors as a therapeutic strategy for TRALI in humans.
Discussion
The pathophysiology of antibody-mediated TRALI involves complex interactions between various blood cells, IgG subclasses, and molecular mediators, rendering difficult the identification of the main molecular and cellular effectors of this pathology. Hence, the availability of murine models of TRALI has permitted a better understanding of the mechanisms implicated in its pathogenesis. The current study describes a new mouse model of antibody-mediated TRALI, based on the expression of a human FcγRIIA/CD32A transgene and the use of a recombinant chimeric human/mouse mAb derived from the anti–MHC I mouse mAb 34-1-2S. Remarkably, the TRALI reactions induced by this recombinant antibody, namely, the early increases in pulmonary elastance, the protein content of BALs, and mortality, were more severe in transgenic animals than in WT animals. Our results indicate that platelet FcγRIIA/CD32A activation exacerbates antibody-mediated TRALI through its involvement in platelet serotonin release and the subsequent activation of 5-HT2A serotonin receptors. These findings provide a rationale for targeting serotonin receptors to attenuate TRALI in humans.
Our data identify FcγRIIA/CD32A as an important determinant of the severity of antibody-mediated TRALI. Nevertheless, a contribution of FcγRI and FcγRIV to TRALI cannot be excluded, given that these receptors have previously been shown to contribute to IgG2a-mediated anaphylaxis.34 In addition, a potential contribution of complement cannot be excluded, as recently highlighted in the case of TRALI-inducing vs TRALI-resistant antibodies.16,35
We and others have previously reported that, in WT mice, monocytes/macrophages are essential to induce TRALI with the mouse IgG2a anti–MHC I mAb 34-1-2S,7,12,13,15 whereas neutrophils and platelets are dispensable.7,8,11,12 Controversies have long existed, however, especially regarding the contribution of the recipient platelets to the pathology, notwithstanding the recent consensus that platelets may not be critically required for the onset and development of TRALI in WT mice.36,37 Here, similar cell-dependent features were observed in CD32A– animals when TRALI was induced by the chimera hIgG1-34-1-2S. In CD32A+ mice, the assessed TRALI responses induced by hIgG1-34-1-2S did not seem to require the presence of circulating neutrophils, showing that expression of FcγRIIA/CD32A in mice does not confer a major additional function on neutrophils during TRALI. Remarkably, liposome clodronate-sensitive cells (monocytes, macrophages, and/or dendritic cells) were not required for the initiation of lung edema in CD32A+ mice, although they contributed to it, indicating a new mechanism and the decisive contribution of another cell type.
The administration of hIgG1-34-1-2S induced thrombocytopenia in WT and CD32A+ mice, probably by 2 different mechanisms. In WT animals, monocyte/macrophage depletion reduced thrombocytopenia, suggesting that some of the anti–MHC I-opsonized platelets were captured by liver and/or splenic macrophages. In contrast, in CD32A+ mice, thrombocytopenia was not affected by macrophage depletion. This effect could result first from an increased adsorption of platelets on the opsonized endothelium, most likely through an FcγRIIA/CD32A–dependent mechanism, as suggested by the enhanced immunohistochemical staining of platelets on lung sections of CD32A+ mice. Second, the increased activation state of these platelets, as revealed by the large amounts of granule-specific molecules released into the blood of CD32A+ mice during TRALI, might stabilize this adsorption. Consequently, in transgenic animals, platelets would be efficiently sequestered on the vessel wall.
The massive release of platelet granule contents also suggests that the amplification of TRALI responses in these animals is platelet dependent. In agreement, prior depletion of circulating platelets abolished the aggravation of TRALI observed in CD32A+ mice. In fact, our analysis identified platelet-derived serotonin as the mediator responsible for FcγRIIA/CD32A–induced aggravation of TRALI, as the latter was abrogated when platelet serotonin stores were pharmacologically depleted in vivo before induction of TRALI. Notably, in CD32A+ mice, low-grade lung edema occurred in the absence of monocytes and macrophages and was completely abolished by fluoxetine treatment, strongly suggesting that FcγRIIA/CD32A–mediated platelet activation and serotonin release are sufficient to initiate the pathogenesis of TRALI.
A major finding of this study was that the 5-HT2A serotonin receptor subtype is responsible for FcγRIIA/CD32A–induced exacerbation of lung edema formation. This property could result from the particular physiology of lung circulation. By targeting 5-HT2A+ vascular smooth muscle cells (VSMCs), serotonin provokes pulmonary arterial vasoconstriction and a subsequent increase in pulmonary arterial pressure.38,39 We previously proposed that the transient increase in pulmonary arterial pressure was responsible for lung edema formation during TRALI.12 This hypothesis was based on several observations: (1) alveolar edema occurs when interstitial edema exceeds a threshold due to plasma leakage through the vessel wall before flooding into the alveoli40 ; (2) during anti-MHC I–induced TRALI, the precapillary arterioles, but not the postcapillary venules, are the exclusive site of edema formation12 ; and (3) NF449, a selective P2X1 receptor antagonist, which blocks VSMC constriction, also inhibits TRALI.12 Similarly, the contraction of pulmonary VSMCs is positively regulated by 5-HT2A serotonin receptors present on their surface38,39 and inhibited by specific 5-HT2 receptor antagonists.41,42 It is therefore tempting to speculate that the abrogation of the increase in pulmonary edema by sarpogrelate might be due to the inhibition of serotonin-induced pulmonary peri-arteriolar VSMC contraction.
Whether the transient increase in pulmonary arterial pressure does occur during the very early steps of TRALI in humans is a question difficult to address because pulmonary arterial pressure is rarely measured during transfusion of a patient. Nevertheless, in 2 cases of human TRALI observed after the transfusion of blood products containing anti-HLA class I antibodies, the first manifestation of the reaction was an increase in pulmonary arterial blood pressure, which occurred before the onset of hypoxemia and acute respiratory distress, while at the same time systemic arterial blood pressure decreased.43,44 These cases point to the possibility that serotonin-induced pulmonary arterial contraction is an early event in the development of TRALI. Of note, whereas serotonin acts as a powerful vasoconstrictor in human pulmonary arteries, it acts as a vasodilator in peripheral vessels.45,46 One may speculate that the vascular territory–dependent effects of serotonin could explain the specific localization of TRALI lesions in pulmonary tissue. Whether the use of selective serotonin reuptake inhibitors in patients with depression is associated with fewer cases of antibody-mediated TRALI is currently unknown.
TRALI is an acute respiratory distress syndrome, the mainstay treatment of which remains general supportive care, including fluid resuscitation or mechanical ventilation.47 No pharmacologic agents able to improve the outcome of acute respiratory distress syndrome have yet been identified. Nevertheless, mouse experiments have suggested that administration of interleukin-10 or C-reactive protein inhibitors could represent promising therapeutic or prophylactic therapies against TRALI.14 Our findings now indicate that administration of sarpogrelate after the onset of TRALI efficiently inhibits edema formation in CD32A+ mice. These results therefore provide a rationale for targeting serotonin receptors to block the pathological progression of TRALI.
Overall, the present humanized model of antibody-mediated TRALI offers new insights into the mechanisms underlying TRALI as it may occur in humans, while identifying a crucial contribution of platelet FcγRIIA/CD32A and serotonin release to the severity of the syndrome. The same mechanism also underlines the role of platelet serotonin in lung pathophysiology, as recently highlighted in IgG-dependent anaphylaxis studies.21,25 This would suggest a similar crucial contribution of platelet FcγRIIA/CD32A to antibody-induced TRALI and IgG-dependent anaphylaxis, through platelet activation and serotonin release, leading to increased vascular permeability and edema formation. Future clinical investigations will aim at determining how our observations can be translated into the human health care setting. In addition to blood transfusion therapy, these findings could have implications in other fields relevant to antibody-mediated damage in which platelets are active players such as organ transplantation and acute rejection.
Acknowledgments
The authors thank M. Freund and her collaborators for animal care, Laurie Ruch and Oceane Schlienger for expert technical assistance, and the National Institutes of Health Tetramer Core Facility (tetramer.yerkes.emory.edu) for provision of MHC-peptide complexes. The authors are particularly grateful to Patrick England and the Plateforme de Biophysique Moléculaire (PFBMI), Institut Pasteur, for help with Bio-layer interferometry measurements, and to J. Mulvihill for revising the English of the manuscript. The visual abstract was created by using https://biorender.com.
This work was supported by a project grant from the Etablissement Français du Sang (APR 2014) and by INSERM, Etablissement Français du Sang, and ARMESA (Association de Recherche et Développement en Médecine et Santé Publique). F.J. is an employee of the CNRS (Centre National de la Recherche Scientifique).
Authorship
Contribution: M.-B.E.M., B.M., S.M., F.T., F.J., and B.H. performed experiments and analyzed data; F.J. contributed essential material; H.d.l.S., B.M., and B.H. designed the research and wrote the manuscript; and C.G. and F.J. critically reviewed the manuscript; and all authors contributed to the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Beatrice Hechler, UMR_S1255 INSERM, Université de Strasbourg, Etablissement Français du Sang-Grand Est, 10 rue Spielmann, BP 36, F-67065 Strasbourg Cedex, France; e-mail: beatrice.hechler@efs.sante.fr.