

Key Points
Inhibition of TNFR2 decreases WBC counts but does not ameliorate hematocrit and splenomegaly in a JAK2-V617F knock-in mouse model.
In a JAK2-V617F knock-in mouse model expressing chimeric TNFR1, anti-human TNFR1 antibody therapy reduces hematocrit and splenomegaly.

Abstract
Chronic nonresolving inflammatory syndrome is a major disease feature in myeloproliferative neoplasms (MPNs). Systemic inflammation promotes the growth of the JAK2-V617F+ hematopoietic stem cell clone and is associated with constitutive symptoms (eg, fever, cachexia, and fatigue). Therefore, it is being discussed whether anti-inflammatory therapy, in addition to the well-established JAK inhibitor therapy, may be beneficial in the control of constitutive symptoms. Moreover, effective control of the inflammatory microenvironment may contribute to prevent transformation into secondary myelofibrosis and acute leukemia. Given the pivotal role of tumor necrosis factor α (TNF-α) in MPN and the distinct roles of TNF-α receptor 1 (TNFR1) and TNFR2 in inflammation, we investigated the therapeutic effects of αTNFR1 and αTNFR2 antibody treatment in MPN-like disease using the JAK2+/VF knock-in mouse model. Peripheral blood counts, bone marrow/spleen histopathology, and inflammatory cytokine levels in serum were investigated. αTNFR2 antibody treatment decreased white blood cells and modulated the serum levels of several cytokines [CXCL2, CXCL5, interleukin-12(p40)], as well as of macrophage colony-stimulating factor, but they lacked efficacy to ameliorate hematocrit and splenomegaly. αTNFR1 antibody treatment resulted in the mild suppression of elevated hematocrit of −10.7% and attenuated splenomegaly (22% reduction in spleen weight). In conclusion, our studies show that TNFR1 and TNFR2 play different roles in the biology of JAK2-V617F–induced disease that may be of relevance in future therapeutic settings.
Introduction
Myeloproliferative neoplasms (MPNs) represent a group of hematologic diseases that exhibit terminal myeloid cell expansion, including erythrocytes, thrombocytes, and leukocytes.1,2 The 3 main Philadelphia chromosome–negative MPN subentities, polycythemia vera (PV), essential thrombocytosis (ET), and primary myelofibrosis (PMF), are characterized by chronic activation of the JAK-STAT pathway resulting from transforming mutations in JAK2, calreticulin, or MPL genes.3-5 Genetic analysis of patients with MPNs revealed that >95% of patients with PV and ∼50% of patients with ET or PMF carry an activating point mutation in the JAK2 gene (JAK2-V617F).6-8 Patients may suffer from a variety of constitutional symptoms, such as fever, cachexia, fatigue, pruritis, night sweats, and C-reactive protein elevation, that are attributed to elevated levels of proinflammatory cytokines.9,10 A major cause of mortality and morbidity is arterial and venous thrombosis. Another common clinical feature, which is most pronounced in PMF, is marked extramedullary hematopoiesis in spleen leading to splenomegaly. Tumor necrosis factor α (TNF-α) is known to be strongly expressed in the different subentities (PV, ET, and PMF), and serum TNF-α levels increase with the allelic ratio of the JAK2-V617F mutation in patients.11 In addition to TNF-α, serum levels of a large number of proinflammatory cytokines, including interleukin-β (IL-1β), IL-6, IL-8, and CXCL10 are chronically elevated in patients with MPN.10,12-17
In bone marrow, malignant and nonmalignant cells, including hematopoietic stem and progenitor cells (HSPCs), monocytes, megakaryocytes, and mesenchymal stromal cells, aberrantly secrete inflammatory cytokines and remodel the bone marrow microenvironment.17-20 In particular, TNF-α, IL-1β, and TIMP promote the growth of the malignant MPN clone, whereas the growth of JAK2 wild-type (WT) cells is suppressed.11,18 It also appears that elevated serum levels of TNF-α and IL-6 are associated with an increased risk for venous thromboembolism.21 Patients with MPN may be treated with phlebotomy, acetylsalicylic acid, hydroxycarbamide, interferon-α (IFN-α), or JAK inhibitors (eg, ruxolitinib) to reduce hematocrit (HCT), splenomegaly, and thrombotic events, as well as to increase overall survival.22 Nevertheless, current therapies often have minor or transient effects only on the inflammatory syndrome and the constitutional symptoms that negatively affect the quality of life in patients with MPN. In patients with myelofibrosis (MF), the pivotal study by Verstovsek and colleagues showed that, upon treatment with ruxolitinib for 28 days, multiple cytokines were reduced in plasma.23 Importantly, upon 24 weeks of therapy with ruxolitinib, reductions in the plasma levels of selected cytokines, including TNF-α, correlated with symptomatic improvements.23 However, another study showed that, upon long-term treatment (>1 month), a number of cytokines were minimally sensitive to ruxolitinib, including CXCL10, IL-1β, IL-5, IL-6, IL-10, IL-16, TNF-α, and VEGF.19 Given the central role of systemic inflammation in MPN development and in disease burden, the current literature debates whether anti-inflammatory therapy, including anticytokine treatment, may be useful to provide more effective suppression of systemic inflammation.17,24-26 This, in turn, may translate into better control of transformation into secondary myelofibrosis and acute leukemia.24,25 In addition, inhibition of the proinflammatory environment in the host may also intervene with the pathophysiology of inflammation-induced secondary neoplasia as skin cancer.27-29
In line with the observation that deletion of TNF-α in a murine transplantation model of MPN attenuated disease development, blockade of the pleiotropic cytokine TNF-α using etanercept, a soluble TNF-α receptor, in patients suffering from MF with myeloid metaplasia, resulted in improvement in constitutional symptoms in 12 of 22 (60%) patients, and 4 patients (20%) showed improvement of cytopenia or spleen size.11,30 It was shown before that TNF-α is important for the expansion of the malignant clone in MPN and that blockade of the TNF-α receptor 2 (TNFR2) pathway decreases clonogenic growth of CD34+ cells isolated from patients with primary MF and of Lin−kit+ bone marrow cells retrovirally transduced to express JAK2-V617F.11,31 However, in vivo blockade of pan–TNF-α did not reduce disease burden in a retrovirally induced JAK2-V617F+ Balb/c bone marrow transplantation model of MPN.31 Therefore, we hypothesized that differential inhibition of the TNF-α receptors TNFR1 and TNFR2 may be more efficacious, because these receptors control opposing cellular signaling pathways and may have divergent effects on disease development.
TNF-α exists in 2 forms, a transmembrane protein (tm-TNF-α) form and a soluble (s-TNF-α) form. Initially, the protein is synthesized as tm-TNF-α. Release from the cell surface and formation of s-TNF-α requires proteolytic cleavage by ADAM17.32 The pleiotropic functions of TNF-α result from binding to 2 distinct receptors. TNFR1 is expressed ubiquitously and binds and is activated by s-TNF-α and tm-TNF-α, whereas TNFR2 is predominantly activated by tm-TNF-α and is expressed only on distinct subsets of cells, such as T- and B-cell types, endothelial cells, different myeloid cell lines, or neurons.33-37 Activation of TNFR1 predominantly triggers proinflammatory and proapoptotic pathways, whereas TNFR2 activation by tm-TNF-α is associated with prosurvival functions in several tissues and activation of regulatory T cells.35 Because of the dichotomic functions of TNFR1 and TNFR2 signaling, blockade of a single TNF-α receptor was shown to exert differential biologic effects compared with complete TNF-α inhibition.34-36 Blockade of TNFR1, but not TNFR2, attenuates disease in multiple sclerosis, rheumatoid arthritis, and a model of chronic inflammation.38-40 Blockade of TNFR2 enhances the efficacy of immunotherapy in mouse models of colon and breast cancer, and TNFR2 was shown to regulate inflammation in models of X-linked proliferative disease 2.41,42
Here, we investigated the therapeutic potential of differential αTNFR1 and αTNFR2 treatment in established MPN-like disease using neutralizing antibodies in the JAK2-V617F knock-in model.
Methods
Mice
JAK2+/VF knock-in mice have been generated and characterized by Mullally et al.43 Floxed heterozygous JAK2+/loxP-VF-loxP C57BL/6 mice were crossed with heterogenic Vav-Cre C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME) to induce expression of the mutated JAK2-V617F kinase in hematopoiesis starting during mouse embryogenesis, as described previously.44 To study the αTNFR1 antibody, JAK2+/VF mice were crossed with a C57BL/6 mouse strain expressing a chimeric TNFR1 consisting of the human extracellular domain and the murine transmembrane and intracellular domains (huTNFR1ecd). This allowed for the treatment of mice with an anti-human antibody isotype H398.45,46 To rule out secondary effects of the Cre recombinase, all JAK2WT control mice were Vav-Cre+.
Antibodies
For αTNFR1 and αTNFR2 therapy, JAK2+/VF mice were treated for 3 weeks. huTNFR1ecd × JAK2+/VF mice received anti-human TNFR1 antibody H398 (20 mg/kg body weight intraperitoneally per injection38,46 ) three times a week. Anti-murine TNFR2 TR75-54.7 antibody was given to JAK2+/VF mice (5 mg/kg body weight intraperitoneally per injection; Bio X Cell, Lebanon, NH) twice a week over 3 weeks.46,47 Control mice received corresponding immunoglobulin G (IgG) isotype controls (αTNFR1: Mouse IgG2a clone C1.18.4; αTNFR2: Armenian Hamster IgG; both from Bio X Cell).
Blood count analysis
Blood was collected weekly during treatment by retrobulbar sinus puncture and measured using an ADVIA 2120 system (Siemens Healthcare, Erlangen, Germany).
Hematoxylin and eosin staining
Paraffin-embedded spleen and bone marrow sections (2 µm) were deparaffinized and rehydrated before staining the tissue in a hematoxylin bath for 10 to 15 minutes. The sections were washed in tap water for 10 to 15 minutes, rinsed in deionized water, and counterstained with eosin for 2 to 3 minutes. Subsequently, the sections were dehydrated in 70% EtOH, 96% EtOH, and EtOHp.a., cleared in Xylol, and mounted. Images were acquired with a Leica DM6000 B microscope and processed using ImageJ software.
Cytokine analysis
Cytokine analysis was performed by Eve Technologies Corporation (Calgary, AB, Canada) using the Mouse Cytokine Array/Chemokine Array 31-Plex kit. For analysis, serum of mice was isolated from peripheral blood after therapy or from untreated JAK2+/VF and JAK2WT mice. Samples were stored at −80°C until measurement.
Statistical analysis
Statistical significance of all experimets was determined using the unpaired 2-tailed Student t test, nonparametric Mann-Whitney U test. A P value < .05 was considered statistically significant (GraphPad Prism v9 software; GraphPad Software Inc., San Diego, CA).
Study approval
Mice were housed under specific pathogen–free conditions in the accredited Animal Research Facility of the Otto-von-Guericke University Medical Faculty. All experiments were performed with approval of the regional government authority Saxony-Anhalt (42502-2-1417). Heparin-anticoagulated blood was drawn after written informed consent was obtained. Experiments were performed according to the guidelines of the Declaration of Helsinki and were approved by the Ethics Committee of the Medical Faculty Magdeburg (protocol MD115/08).
Results
The effects of TNFR2 blockade on MPN-like disease and on serum cytokine/chemokine levels
We investigated the consequences of αTNFR2 antibody treatment in JAK2+/VF knock-in mice43 upon challenge with an anti-murine TNFR2 antibody (TR75-54.7) to analyze the effects of single TNFR2 blockade on MPN disease burden (Figures 1-3). To demonstrate pathway inhibition, isolated splenocytes from JAK2+/VF mice were incubated with increasing antibody concentrations and then stimulated with TNF-α. This resulted in an αTNFR2 concentration-dependent decrease in the phosphorylation of p65 NF-κB and p38 in murine JAK2-V617F+ splenocytes (supplemental Figure 6A). αTNFR2 treatment had only a minor impact on elevated HCT and hemoglobin (HGB) levels in JAK2+/VF mice. The mean HCT declined only marginally in αTNFR2-treated mice (−4.8% over 3 weeks) with a similar effect in IgG-treated mice (−3.0% over 3 weeks) (Figure 1A). Compared with untreated JAK2+/VF mice, end point HCT levels of treated mice remained high (76.3% untreated, 78.8% αTNFR2, 82.3% IgG), indicating no improvement in this parameter of disease burden (Figure 1A-B). Mean HGB in αTNFR2- and IgG-treated mice remained unchanged during treatment and was similar to untreated JAK2+/VF mice (Figure 1C). In line with these results, hepcidin levels of αTNFR2- or IgG-treated mice were similar to untreated JAK2+/VF mice, but they were decreased compared with JAK2WT mice (supplemental Figure 1A).48 Interestingly, platelet (PLT) and white blood cell (WBC) counts declined during treatment. This change was rather small for PLT counts, with a decrease from 1152 × 109/L to 916 × 109/L upon αTNFR2 treatment (Figure 1E). However, the WBC count decreased significantly from 13.0 to 9.3 × 109/L during treatment, reaching a level comparable to that observed in JAK2WT mice (9.3 × 109/L) (Figure 1D). Of note, splenomegaly was not reduced by αTNFR2 treatment, and mean weights of spleens from αTNFR2- or IgG-treated mice were similar to untreated JAK2+/VF mice (537, 530, and 589 mg, respectively) (Figure 1G-H). In addition, there was no effect of αTNFR2 and the corresponding IgG treatment on splenic histopathology in JAK2+/VF mice, and there were no apparent changes in bone marrow histology (Figure 2). T- and B-cell numbers were similar in the peripheral blood from αTNFR2- and IgG-treated mice (1.883 × 109/L and 1.801 × 109/L vs 3.151 × 109/L and 2.750 × 109/L in αTNFR2- and IgG-treated mice, respectively; supplemental Figure 3C-D). Similar to the bone marrow analysis of Mullally et al,43 the number of LK cells (Lin−, ckit+, Sca1−; supplemental Figure 4C), especially megakaryocyte-erythrocyte progenitors (MEPs), was significantly increased in the spleen of untreated JAK2+/VF mice compared with JAK2WT mice; this was primarily an effect of the increase in MEP cells in JAK2+/VF mice. Cell numbers were similarly reduced by αTNFR2 and IgG treatment without any statistically significant changes (supplemental Figure 4A).
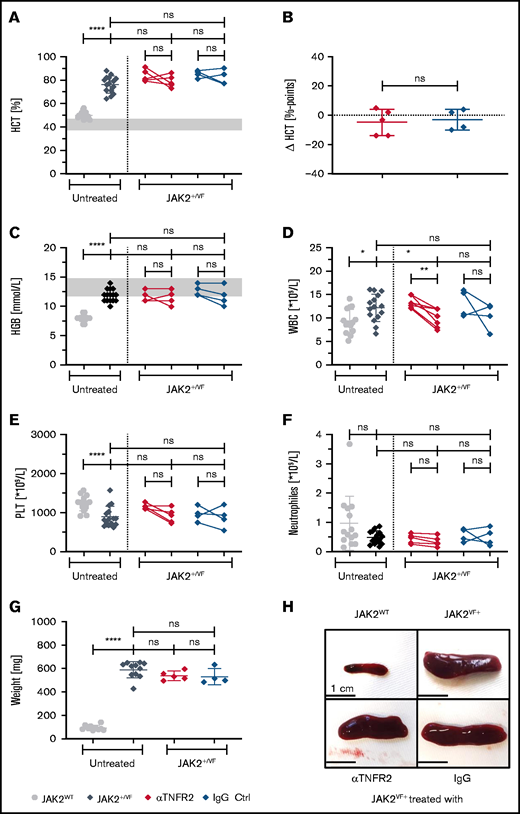
MPN disease parameters upon 3 weeks of treatment using an αTNFR2 antibody. JAK2+/VF mice were treated intraperitoneally, 5.0 mg/kg body weight, 2 times a week for 3 weeks with an anti-mouse TNFR2 antibody (clone TR75-54.7) or polyclonal Armenian Hamster IgG as control antibody. Blood counts were analyzed weekly during the treatment phase. All mice were 12 to 13 weeks old at the end of therapy JAK2WT and JAK2+/VF mice were age matched (11-15 weeks). (A-F) HCT and blood count analysis of untreated JAK2+/VF mice (n = 15), JAK2WT mice (n = 14), and JAK2+/VF mice treated with αTNFR2 antibody (n = 5) or IgG (n = 4). (B) Change in HCT (ΔHCT) in αTNFR2-treated mice and corresponding control group. Differences between HCT at the start and end of treatment in αTNFR2-treated mice and IgG-treated controls (ΔHCT = HCTEnd − HCTStart). (G) Spleen weights for untreated JAK2+/VF mice (n = 11), JAK2WT mice (n = 10), and JAK2+/VF mice treated with αTNFR2 antibody (n = 5) or IgG (n = 4). (H) Representative photographs of spleens from untreated control mice (n = 3) and JAK2+/VF mice (n = 6) treated with αTNFR2 antibody (n = 2) or IgG (n = 2). *P < .05, **P < .01, ***P < .001 ****P < .0001. Ctrl, control; ns, not significant.
MPN disease parameters upon 3 weeks of treatment using an αTNFR2 antibody. JAK2+/VF mice were treated intraperitoneally, 5.0 mg/kg body weight, 2 times a week for 3 weeks with an anti-mouse TNFR2 antibody (clone TR75-54.7) or polyclonal Armenian Hamster IgG as control antibody. Blood counts were analyzed weekly during the treatment phase. All mice were 12 to 13 weeks old at the end of therapy JAK2WT and JAK2+/VF mice were age matched (11-15 weeks). (A-F) HCT and blood count analysis of untreated JAK2+/VF mice (n = 15), JAK2WT mice (n = 14), and JAK2+/VF mice treated with αTNFR2 antibody (n = 5) or IgG (n = 4). (B) Change in HCT (ΔHCT) in αTNFR2-treated mice and corresponding control group. Differences between HCT at the start and end of treatment in αTNFR2-treated mice and IgG-treated controls (ΔHCT = HCTEnd − HCTStart). (G) Spleen weights for untreated JAK2+/VF mice (n = 11), JAK2WT mice (n = 10), and JAK2+/VF mice treated with αTNFR2 antibody (n = 5) or IgG (n = 4). (H) Representative photographs of spleens from untreated control mice (n = 3) and JAK2+/VF mice (n = 6) treated with αTNFR2 antibody (n = 2) or IgG (n = 2). *P < .05, **P < .01, ***P < .001 ****P < .0001. Ctrl, control; ns, not significant.
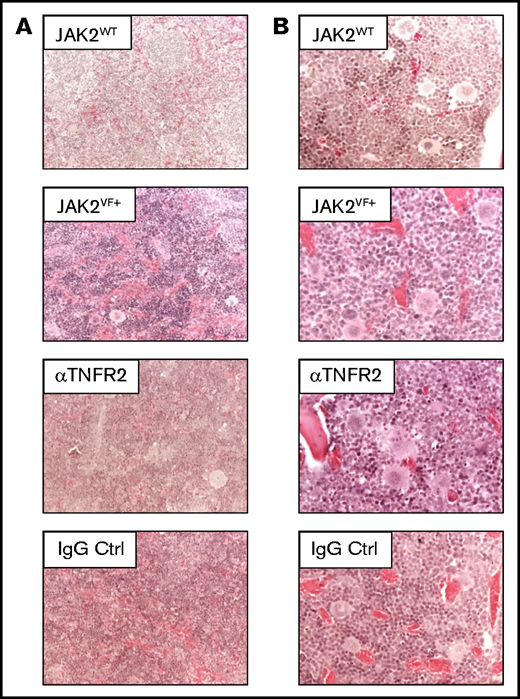
Splenic and bone marrow structure in JAK2+/VF mice after αTNFR2 treatment. (A) Representative pictures of hematoxylin and eosin–(H&E)-stained spleen sections from untreated JAK2WT(n = 1), untreated JAK2VF mice (n = 2) and JAK2+/VF mice treated with αTNFR2 antibody (n = 3) or IgG (n = 3); original magnification ×200. (B) Representative photomicrographs of H&E-stained bone marrow sections from untreated JAK2WT (n = 1), untreated JAK2VF mice (n = 2) and JAK2+/VF mice treated with αTNFR2 antibody (n = 3) or IgG (n = 3); original magnification ×400. Ctrl, control.
Splenic and bone marrow structure in JAK2+/VF mice after αTNFR2 treatment. (A) Representative pictures of hematoxylin and eosin–(H&E)-stained spleen sections from untreated JAK2WT(n = 1), untreated JAK2VF mice (n = 2) and JAK2+/VF mice treated with αTNFR2 antibody (n = 3) or IgG (n = 3); original magnification ×200. (B) Representative photomicrographs of H&E-stained bone marrow sections from untreated JAK2WT (n = 1), untreated JAK2VF mice (n = 2) and JAK2+/VF mice treated with αTNFR2 antibody (n = 3) or IgG (n = 3); original magnification ×400. Ctrl, control.
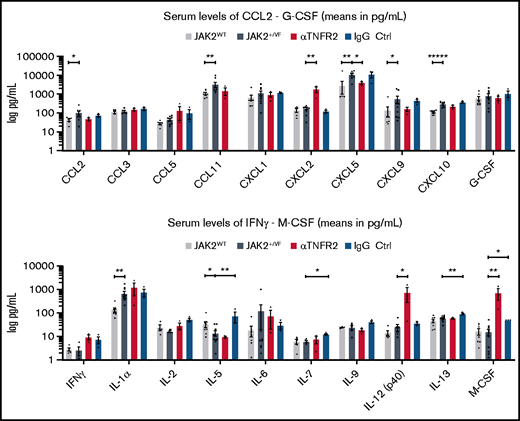
Cytokine expression in JAK2+/VF mice upon αTNFR2 treatment. Comparison of serum concentrations of cytokines in untreated JAK2+/VF mice, untreated JAK2WT mice, αTNFR2 antibody–treated JAK2+/VF mice, and IgG-treated JAK2+/VF mice, as analyzed by Eve Technologies Corporation using a Mouse Cytokine Array/ Chemokine Array 31-Plex. Cytokine concentrations in serum. From left to right: untreated JAK2WT (n = 4-5), untreated JAK2+/VF (n = 4-10), αTNFR2 antibody treated JAK2+/VF (n = 3) and IgG treated JAK2+/VF mice (n = 3) in serum. Cytokines are depicted in alphabetical order. For statistical analysis, values “red flagged” as “out of range” or “extrapolated values” by Eve Technologies were excluded from the presented data set. Cytokine concentrations are only presented, if the data set of each cytokine consisted of n (values) ≥ 3 after exclusion of “out of range” and “extrapolated” values. *P < .05, **P < .01, ****P < .0001.
Cytokine expression in JAK2+/VF mice upon αTNFR2 treatment. Comparison of serum concentrations of cytokines in untreated JAK2+/VF mice, untreated JAK2WT mice, αTNFR2 antibody–treated JAK2+/VF mice, and IgG-treated JAK2+/VF mice, as analyzed by Eve Technologies Corporation using a Mouse Cytokine Array/ Chemokine Array 31-Plex. Cytokine concentrations in serum. From left to right: untreated JAK2WT (n = 4-5), untreated JAK2+/VF (n = 4-10), αTNFR2 antibody treated JAK2+/VF (n = 3) and IgG treated JAK2+/VF mice (n = 3) in serum. Cytokines are depicted in alphabetical order. For statistical analysis, values “red flagged” as “out of range” or “extrapolated values” by Eve Technologies were excluded from the presented data set. Cytokine concentrations are only presented, if the data set of each cytokine consisted of n (values) ≥ 3 after exclusion of “out of range” and “extrapolated” values. *P < .05, **P < .01, ****P < .0001.
To determine whether αTNFR2 antibody treatment results in suppression of proinflammatory cytokines in vivo, a panel of cytokines was monitored in serum (Figure 3; supplemental Table 1) utilizing the commercially available Mouse Cytokine Array/Chemokine Array 31-Plex service (Eve Technologies). The analysis of serum cytokine concentrations confirmed strong differences between JAK2WT and JAK2+/VF mice (Figure 3). A number of cytokines (CCL2, CCL11, CXCL5, CXCL9, CXCL10, and IL-1α) were significantly upregulated compared with JAK2WT mice (Figure 3; supplemental Table 1). In particular, the 3.79-fold increase in CXCL5 (JAK2WT vs JAK2+/VF: 2860 pg/mL vs 10 854 pg/mL) and the 2.70-fold increase in CXCL10 (JAK2WT vs JAK2+/VF: 110.7 pg/mL vs 299.3 pg/mL) recapitulate the expression pattern observed in patients with MPN.12,49 Interestingly, despite the apparent lack of efficacy to ameliorate hematologic disease parameters, treatment with αTNFR2 antibody decreased the concentration of a group of cytokines (CCL2, CCL11, CXCL5, or CXCL9) to levels similar to those found in JAK2WT mice (Figure 3; supplemental Table 1), which was not observed in IgG-treated mice. Prototypic proinflammatory cytokines, such as CCL5, IFN-γ, and CXCL2, were increased by αTNFR2 treatment (3.01-fold, 3.59-fold, and 10.53-fold increase, respectively), whereas CCL3, IL-2, and IL-1α basically remained unchanged or were upregulated only slightly by TNFR2 blockade (1.25-fold, 1.73-fold, and 1.83-fold change, respectively). Of note, monocyte/macrophage colony-stimulating factor was significantly upregulated 46.51-fold (supplemental Table 1). The changes in serum cytokine levels observed upon administration of high-dose IgG (control-isotype antibody) are not unexpected. There is a large body of literature showing that high-dose IgG downregulates proinflammatory cytokines (recently reviewed by Liu et al).50 In addition, high-dose IgG is a well-established immunomodulatory therapy in humans. The underlying molecular mechanisms of IgG effectiveness were described to be conferred by the 2 functional domains of IgG, the F(ab)′2 fragment and the Fc fragment, which are responsible for FcR and complement binding. Downregulation of proinflammatory cytokines is believed to be mediated through the Fc-mediated mechanisms on endothelial cells, dendritic cells, and macrophages.50 Thus, our results on the modulation of proinflammatory serum cytokine levels by high-dose IgG are consistent with the current literature.
αTNFR1 antibody treatment attenuates features of MPN-like disease in the JAK2-V617F mouse model
Next, we investigated the effects of TNFR1 blockade in JAK2+/VF mice.43 JAK2+/VF mice were crossed with huTNFR1ecd mice, as described.40 Because human TNFR1 is activated by mouse TNF-α, using this model allowed us to evaluate the previously characterized anti-human TNFR1 antibody H398 for therapeutic activity.40
As described for the JAK2+/VF knock-in mouse model, huTNFR1ecd × JAK2+/VF mice developed a PV-like phenotype with highly increased HCT and HGB, elevated WBC counts, and prominent splenic extramedullary hematopoiesis resulting in splenomegaly (Figures 4 and 5).43 αTNFR1 antibody treatment 3 times per week for 3 weeks was well tolerated without apparent changes in the behavior or appearance of mice. Unlike αTNFR2, the αTNFR1 antibody schedule used reduced the mean HCT from 76.1% to 65.4% (red; Figure 4A). This change was not observed in isotype IgG–treated control mice; the mean HCT remained stable during treatment (71.1-73.8%; blue; Figure 4A). The change in HCT percentage points between αTNFR1-treated mice and IgG-treated mice was statistically significant (Figure 4B). A similar change was observed when assessing HGB levels (Figure 4C); there was a general decline in HGB (from a mean of 11.0 mmol/L to 10.0 mmol/L) in αTNFR1-treated mice, whereas IgG-treated mice experienced a general increase (from a mean of 10.2 mmol/L to 10.9 mmol/L). Hepcidin levels were strongly reduced in huTNFR1ecd × JAK2+/VF mice compared with WT controls. Treatment of huTNFR1ecd × JAK2+/VF mice with αTNFR1 or IgG control antibodies did not change hepcidin levels compared with untreated huTNFR1ecd × JAK2+/VF mice, indicating no improvement in the disturbed iron metabolism observed in the PV-like disease (supplemental Figure 1B).48 PLT counts were not altered significantly in αTNFR1- and IgG-treated mice, with strong variations in individual mice. However, nearly all counts measured in individual mice were in the range of normal values (766-1657 × 109/L) (Figure 4E). The WBC count was increased by the JAK2-V617F mutation, as reported previously (Figure 4D).43,51 In contrast to TNFR2 blockade, the elevated WBC count in huTNFR1ecd × JAK2+/VF mice was upregulated even further by αTNFR1 therapy (from a mean of 13.5 × 109/L to 16.1 × 109/L) (Figure 4D). In isotype IgG–treated JAK2+/VF mice, the mean WBC count remained unchanged during treatment, resulting in a strong difference between the 2 groups at the end of treatment (αTNFR1: 16.1 × 109/L; IgG: 11.8 × 109/L). Although the WBC count is affected by the JAK2-V617F mutation, the number of neutrophils was similar in all groups and did not change during αTNFR1 or IgG treatment (Figure 4F). Changes in mean corpuscular volume (MCV) and red blood cell (RBC) counts are depicted in supplemental Figure 2C and D. Interestingly, upon αTNFR1 antibody treatment, there was a 22% reduction in mean spleen weight from 630 mg to 492 mg (Figure 4G); however, IgG treatment also resulted in a reduction of 17% (mean spleen weight: 520 mg). This may be induced, in part, by an anti–TNF-α and anti–IL-1-mediated molecular/cellular immunomodulatory mechanism that has been described with treatment with high-dose IgG.52-55 In addition, the migration of human JAK2-V617F+ granulocytes along a CXCL12/TNF-α gradient was decreased significantly by TNFR1 inhibition (CXCL12/TNFα: 34 338 ± 7065 migrating cells; CXCL12/TNFα + αTNFR1: 19 094 ± 3816 migrating cells). TNFR2 blockade did not result in any change in migration (CXCL12/TNFα + αTNFR2: 34 604 ± 8164 migrating cells) (supplemental Figure 5).
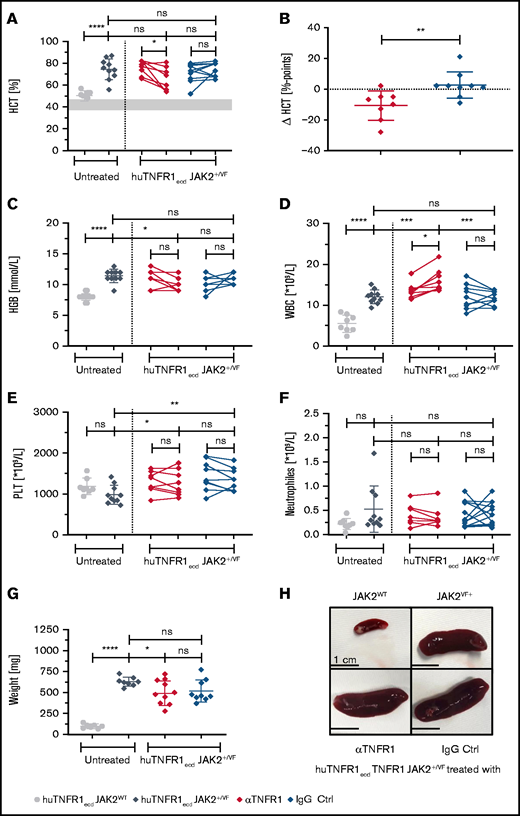
MPN disease parameters upon 3 weeks of treatment using an αTNFR1 antibody. huTNFR1ecd × JAK2+/VF mice were treated intraperitoneally, 20 mg/kg body weight, 3 times a week for 3 weeks with an anti-human TNFR1 antibody (clone H398; Institut fur Zellbiologie un Immunologie Stuttgart) or an IgG2a isotype control Ab (clone C1.18.4). Blood counts were analyzed weekly during the treatment phase. All antibody-treated huTNFR1ecd × JAK2+/VF mice were 12 or 13 weeks old at the end of therapy; huTNFR1ecd × JAK2WT mice and huTNFR1ecd × JAK2+/VF mice were age matched (11-15 weeks). (A-F) HCT and blood count analysis of untreated huTNFR1ecd × JAK2+/VF mice (n = 10) and JAK2WT mice (n = 8) and huTNFR1ecd × JAK2+/VF mice treated with αTNFR1 antibody (n = 8) or IgG (n = 9). (B) Change in HCT (ΔHCT) in αTNFR1-treated mice and corresponding control group. Differences between HCT at the start and end of the treatment phase in αTNFR1-treated mice and IgG control mice (Δ HCT = HCTEnd − HCTStart). (G) Spleen weights for untreated huTNFR1ecd × JAK2+/VF mice (n = 8) and JAK2WT mice (n = 7) and huTNFR1ecd × JAK2+/VF mice treated with αTNFR1 antibody (n = 9) or IgG (n = 9). (H) Representative photographs of spleens from untreated huTNFR1ecd × JAK2WT mice (n = 3) and huTNFR1ecd × JAK2+/VF mice (n = 3) and huTNFR1ecd × JAK2+/VF mice treated with αTNFR1 antibody (n = 5) or IgG (n = 5). *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, not significant.
MPN disease parameters upon 3 weeks of treatment using an αTNFR1 antibody. huTNFR1ecd × JAK2+/VF mice were treated intraperitoneally, 20 mg/kg body weight, 3 times a week for 3 weeks with an anti-human TNFR1 antibody (clone H398; Institut fur Zellbiologie un Immunologie Stuttgart) or an IgG2a isotype control Ab (clone C1.18.4). Blood counts were analyzed weekly during the treatment phase. All antibody-treated huTNFR1ecd × JAK2+/VF mice were 12 or 13 weeks old at the end of therapy; huTNFR1ecd × JAK2WT mice and huTNFR1ecd × JAK2+/VF mice were age matched (11-15 weeks). (A-F) HCT and blood count analysis of untreated huTNFR1ecd × JAK2+/VF mice (n = 10) and JAK2WT mice (n = 8) and huTNFR1ecd × JAK2+/VF mice treated with αTNFR1 antibody (n = 8) or IgG (n = 9). (B) Change in HCT (ΔHCT) in αTNFR1-treated mice and corresponding control group. Differences between HCT at the start and end of the treatment phase in αTNFR1-treated mice and IgG control mice (Δ HCT = HCTEnd − HCTStart). (G) Spleen weights for untreated huTNFR1ecd × JAK2+/VF mice (n = 8) and JAK2WT mice (n = 7) and huTNFR1ecd × JAK2+/VF mice treated with αTNFR1 antibody (n = 9) or IgG (n = 9). (H) Representative photographs of spleens from untreated huTNFR1ecd × JAK2WT mice (n = 3) and huTNFR1ecd × JAK2+/VF mice (n = 3) and huTNFR1ecd × JAK2+/VF mice treated with αTNFR1 antibody (n = 5) or IgG (n = 5). *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, not significant.
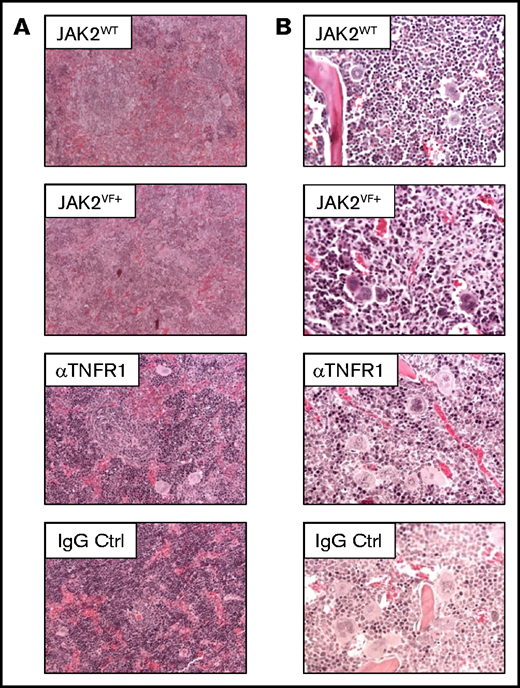
Splenic and bone marrow structure in huTNFR1ecd × JAK2+/VF mice after αTNFR1 treatment. (A) Representative photomicrographs of hematoxylin and eosin (H&E)-stained spleen sections from untreated huTNFR1ecd × JAK2+/VF mice (n = 3) and JAK2WT mice (n = 2) and huTNFR1ecd × JAK2+/VF mice treated with αTNFR1 antibody (n = 2) or IgG (n = 2); original magnification ×200. (B) Representative microphotographs of H&E-stained bone marrow sections from untreated huTNFR1ecd × JAK2+/VF mice (n = 3) and JAK2WT mice (n = 2) and huTNFR1ecd × JAK2+/VF mice treated with αTNFR1 antibody (n = 2) or IgG (n = 2); original magnification ×400.
Splenic and bone marrow structure in huTNFR1ecd × JAK2+/VF mice after αTNFR1 treatment. (A) Representative photomicrographs of hematoxylin and eosin (H&E)-stained spleen sections from untreated huTNFR1ecd × JAK2+/VF mice (n = 3) and JAK2WT mice (n = 2) and huTNFR1ecd × JAK2+/VF mice treated with αTNFR1 antibody (n = 2) or IgG (n = 2); original magnification ×200. (B) Representative microphotographs of H&E-stained bone marrow sections from untreated huTNFR1ecd × JAK2+/VF mice (n = 3) and JAK2WT mice (n = 2) and huTNFR1ecd × JAK2+/VF mice treated with αTNFR1 antibody (n = 2) or IgG (n = 2); original magnification ×400.
Histology of αTNFR1- and IgG-treated mice did not reveal apparent changes in splenic and bone marrow histopathology (Figure 5). Colony-forming unit for granulocytes and macrophages and burst-forming unit assays were performed using lineage negative (Lin−Kit+) bone marrow cells. This study shows that there is indeed a trend toward a TNFR2-dependent inhibition of colony-forming unit for granulocytes and macrophages; however, in comparison with the data reported by Heaton and colleagues31 the difference in clonogenic growth observed is small (supplemental Figure 3A-B). Most likely, this is due to differences in the mouse models used. Heaton et al used a retrovirally induced JAK2-V617F+ Balb/c bone marrow transplantation model featuring high TNF-α levels. Treatment with αTNFR1 had no effect on peripheral blood T- or B-cell numbers compared with IgG treatment (supplemental Figure 3E-F). As expected, the composition of HSPCs in the spleen of huTNFR1ecd × JAK2+/VF mice is similar to JAK2+/VF mice. LK cells are increased in huTNFR1ecd × JAK2+/VF mice compared with huTNFR1ecd × JAK2+/+ mice, with a strong increase in MEP cells in this compartment. Treatment with αTNFR1 or IgG did not appear to affect the composition of HSPCs (supplemental Figure 4B).
Discussion
Classic Philadelphia chromosome–negative MPNs serve as a paradigm for the concept of oncoinflammation.16,17,25,26 Somatic mutations arising in the neoplastic clone sustain a chronic inflammatory state that results from the aberrant production of inflammatory soluble mediators released from malignant and nonmalignant cells.10,13,20,23 Cellular contributors to inflammation are HSPCs, monocytes, megakaryocytes, and mesenchymal stromal cells (for a review, see Jutzi and Mullaly17 ).
Among other soluble inflammatory factors, such as IL-1β and TIMP, TNF-α has been shown to play a pivotal role in MPN-induced systemic inflammation, because it promotes clonal expansion of JAK2-V617F+ cells.11,18 TNF-α expression is increased in hematopoietic stem cells, multipotent progenitor cells, common lymphoid progenitor cells, monocytes, and mature B cells from patients with MF.31 A similar result was found in hematopoietic stem cells, multipotent progenitor cells, and MEPs isolated from bone marrow from Balb/c mice with MPN induced by transplantation of donor cells transduced with the mutated JAK2-V617F gene.31 Interestingly, defective negative regulation of Toll-like receptor signaling via IL-10 leads to excessive TNF-α production in monocytes from patients with MPNs.56 Nevertheless, there are several publications indicating the importance of inflammation and of TNF-α in particular on MPN development. In this regard, it has been shown that treatment with pan–TNF-α inhibitors does not reduce disease development or pathologic parameters in murine MPN.31 The investigators concluded that differential signaling from TNFR1 and/or TNFR2 underlies the competitive advantage of MPN cells. Indeed, Heaton et al investigated the role of TNFR1 and TNFR2 in clonogenic assays of CD34+ cells isolated from patients with MF and, using bone marrow cells, retrovirally transduced to express JAK2-V617F.31 They described an autocrine feedback loop to escape apoptosis through the TNF-α–TNFR2 pathway. Anti-TNFR2 treatment selectively suppressed myeloid colony formation of CD34+ cells isolated from patients with MF and of murine Lin−Kit+ bone marrow cells. Here, we extended these studies to in vivo treatment and comprehensively analyzed the differential role of TNF-α receptors TNFR1 and TNFR2 in hematologic disease parameters and proinflammatory cytokine levels.
Upon TNFR2 blockade, there was a striking reduction in the WBC count. Other parameters of the blood count, in particular factors defining PV (HCT, HGB, and RBC), remained unchanged upon αTNFR2 treatment. There also was no effect on splenomegaly or on splenic histopathology. Importantly, the cytokine signature of serum levels elevated in JAK2-V617F knock-in mice described here recapitulates the signature found in myelofibrotic 6-month-old JAK2-V617F × Vav-Cre knock-in mice.20,43 Thus, similar to age-matched JAK2WT controls, levels of CCL2, CCL11, CXCL5, CXCL9, CXCL10, and IL-1α were upregulated in the 11- to 15-week-old JAK2+/VF mice used in this study. Although IL-9 and CCL3 were highly elevated in the myelofibrotic stage, in the early stage of disease (as described in this study) their levels were unchanged compared with WT controls. Thus, it seems that IL-9 and CCL3 may play a role in the development of MF. Interestingly, the expression of CXCL2, IL-12(p40), and monocyte/macrophage colony-stimulating factor expression were significantly upregulated by TNFR2 inhibition: 10.5-fold, 29.9-fold, and 46.5-fold, respectively. This is of particular interest because CXCL2 is secreted by mast cells and macrophages, and it functions as a major player in control of the early stage of neutrophil recruitment during tissue inflammation.57 CXCL5 was significantly downregulated by TNFR2 inhibition to the levels observed in JAK2WT mice. IgG treatment also modulated, in part, the cytokine levels of JAK2+/VF mice. CCL2 and CXCL2 were downregulated compared with untreated mice. These changes in serum cytokine levels upon administration of high-dose IgG (control isotype antibody) were not unexpected, because there are numerous reports of downregulation of proinflammatory cytokines upon high-dose IgG treatment. For a review please see Liu et al.50
Crossing of JAK2+/VF mice with huTNFR1ecd mice40 allowed to evaluate the previously described anti-human TNFR1 antibody H398.38,40,46 Treatment of huTNFR1ecd × JAK2+/VF mice with the H398 antibody led to a significant reduction in HCT (10.7% points). The decrease in HCT is in a comparable range as observed in JAK2+/VF mice treated with ruxolitinib (60 mg/kg body mass twice daily for 9 days), but it failed to reach the normal levels observed in JAK2WT mice.58 In contrast to αTNFR2 treatment, WBC count was increased by αTNFR1 treatment. This effect may be explained by a shift from the extravascular compartment to peripheral blood induced by αTNFR1 blockade. In addition, upon αTNFR1 antibody treatment, there was a 22% reduction in mean spleen weight. This might be explained, in part, by the reduction in granulocyte migration along the important CXCL12/CXCR4 pathway, which plays a pivotal role in the development of splenomegaly in JAK2-V617F+ MPN.59-62 However, treatment using isotype IgG control also reduced the mean spleen weight by 17%, indicating a positive effect of nonspecific IgG treatment on splenomegaly in this MPN model. This is consistent with recent publications describing the impact of nonspecific IgG treatment on different types of cancer in mouse models and chronic inflammation in different disease settings in humans.52,63
Overall, we show dichotomic functions of TNFR1 and TNFR2 in the biology of JAK2-V617F–induced disease. It is tempting to speculate that differential anti-TNFR1 and anti-TNFR2 treatment could be beneficial in patients with MPN. Thus, TNFR1 inhibition may decrease HCT and splenomegaly, whereas TNFR2 inhibition may reduce leukocytosis. The mouse anti-human TNFR1 antibody H398 that was used in this study has recently been humanized64 and would be available for clinical evaluation. Patients with high-risk primary and secondary MF exhibiting highly symptomatic disease and at high risk for transformation into acute leukemia may be a suitable study population to explore differential αTNFR1/2 therapy, with or without JAK inhibition or other drugs, depending on poorly controlled disease parameters.
Acknowledgments
The authors thank Stephanie Adam-Frey, Anja Sammt, and Corinna Fahldieck for excellent technical support and animal care, as well as Radek Skoda and Jan Stetka (University of Basel, Basel, Switzerland) for advice and discussions on an earlier version of this manuscript.
This work was supported by the German Research Foundation (DFG; SFB854, project A20), by the Europäischer Fonds für Regionale Entwicklung (European Regional Development Fund) (Autonomie im Alter: PhytoHäm, 2019-21) and by the German Ministry of Education and Research (BMBF; e.bio JAK-Sys) (all to T.F.).
Authorship
Contribution: P.M., C.K.B., and T.R.H. performed experiments and analyzed data; A.M.W., F.R., and K.P. provided essential reagents; P.M. and T.F. designed the research and analyzed data; and all authors wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Thomas Fischer, Department of Hematology and Oncology, Center for Internal Medicine, Otto-von-Guericke University Medical Center, Leipzigerstr. 44, 39120 Magdeburg, Germany; e-mail: thomas.fischer@med.ovgu.de.

