

Key Points
Patient-specific NGS-MRD monitoring using non-DTA mutations after alloHCT is independently prognostic for relapse and survival.
The kinetics rather than a single time point should be further evaluated when DTA mutations are used for MRD monitoring after alloHCT.

Abstract
Next-generation sequencing (NGS)-based measurable residual disease (MRD) monitoring in patients with acute myeloid leukemia (AML) is widely applicable and prognostic prior to allogeneic hematopoietic cell transplantation (alloHCT). We evaluated the prognostic role of clonal hematopoiesis–associated DNMT3A, TET2, and ASXL1 (DTA) and non-DTA mutations for MRD monitoring post-alloHCT to refine MRD marker selection. Of 154 patients with AML, 138 (90%) had at least one mutation at diagnosis, which were retrospectively monitored by amplicon-based error-corrected NGS on day 90 and/or day 180 post-alloHCT. MRD was detected in 34 patients on day 90 and/or day 180 (25%). The rate of MRD positivity was similar when DTA and non-DTA mutations were considered separately (17.6% vs 19.8%). DTA mutations had no prognostic impact on cumulative incidence of relapse, relapse-free survival, or overall survival in our study and were removed from further analysis. In the remaining 131 patients with at least 1 non-DTA mutation, clinical and transplantation-associated characteristics were similarly distributed between MRD-positive and MRD-negative patients. In multivariate analysis, MRD positivity was an independent adverse predictor of cumulative incidence of relapse, relapse-free survival, and overall survival but not of nonrelapse mortality. The prognostic effect was independent of different cutoffs (above limit of detection, 0.1% and 1% variant allele frequency). MRD log-reduction between diagnosis and post-alloHCT assessment had no prognostic value. MRD status post-alloHCT had the strongest impact in patients who were MRD positive prior to alloHCT. In conclusion, non-DTA mutations are prognostic NGS-MRD markers post-alloHCT, whereas the prognostic role of DTA mutations in the posttransplant setting remains open.
Introduction
Allogeneic hematopoietic cell transplantation (alloHCT) is the most potent consolidation therapy for patients with acute myeloid leukemia (AML).1,2 It represents the standard treatment of high-risk or relapsed AML patients.1,2 Nevertheless, relapse occurs in 25% to 55% of patients with AML undergoing alloHCT.3 The 2-year overall survival (OS) of relapsed patients after alloHCT ranges between 14% and 25%, with even fewer of them achieving a long-term cure. Thus, optimizing outcome of alloHCT by reducing relapse rates is urgently needed. Detecting molecular relapse before clinical relapse offers the opportunity for early interventions. In addition to approaches that aim for a reinforcement of the graft-versus-leukemia effect (eg, donor lymphocyte infusions or reduction of immunosuppression),4,5 other therapeutic strategies are directly targeting the leukemic cell and are currently being evaluated in clinical trials.6,7 Due to emerging clinical implications of molecular relapse, measurable residual disease (MRD) monitoring post-alloHCT has been recommended as routine follow-up for patients undergoing transplant.8 However, many challenges remain in this setting.9 Requirements for the ideal MRD method not only include a high level of sensitivity (at least 10−3)10 and specificity for relapse prediction but also the ability to be widely (eg, for all patients) and easily (eg, based on peripheral blood) applicable. Furthermore, the technique itself, as well as the interpretation of results, should be easy to be standardized. In this context, next-generation sequencing (NGS)-based error-corrected sequencing is a promising MRD approach.10-13 NGS-based MRD has shown robust results in patients with AML before alloHCT.14-20 Here, a cumulative incidence of relapse (CIR) of 66% in MRD-positive patients vs 17% in MRD-negative patients at 5 years was identified.14 However, NGS-based MRD is not well studied in patients after alloHCT.
Previous studies have shown that mutations in the clonal hematopoiesis (CH)-associated genes DNMT3A, TET2, and ASXL1 (DTA)21,22 did not discriminate relapse risk in AML patients who were in remission.13,15 It is conceivable that reappearance of recipient cells after alloHCT indicates relapse and that DTA mutations may be prognostic after alloHCT.23 In the current study, we aimed to evaluate in a large cohort of AML patients an error-corrected NGS-based MRD amplicon approach on day 90 and day 180 after alloHCT for its applicability and prognostic impact.
Patients, materials, and methods
Patients
Patients with AML (excluding acute promyelocytic leukemia) aged >18 years were included who underwent alloHCT between 1998 and 2017 at Hannover Medical School. Requirement for inclusion in this retrospective cohort study was availability of DNA at diagnosis as well as on day 90 and/or day 180 after alloHCT. Patients were excluded if they relapsed or died within the first 90 days after alloHCT (14% of the patients in our database) or had <1 year clinical follow-up. Within these criteria, 154 patients were evaluated by NGS with a myeloid panel on the Illumina platform for mutations at time of diagnosis. Of 154 initial patients, 14 patients had no mutation, and the NGS-MRD assay failed in 2 patients. Patients with and without mutations at diagnosis had similar patient characteristics (data not shown) and underwent transplant in a similar time period (with mutation: median time of diagnosis, October 2013 [range, January 1998-April 2017]; no mutation: median time of diagnosis, June 2013 [range, September 1996-March 2016]). Thus, 138 patients were included in the final cohort (supplemental Figure 1). Of those, 47 patients underwent myeloablative conditioning (MAC; 34%) and 91 patients received reduced-intensity conditioning (RIC; 66%) before alloHCT.
The scheme for clinical follow-up generally was once weekly until day 100 after alloHCT, once every 2 weeks until day 180, once per month until the end of the first year, once every 3 months during years 2 and 3, once every 6 months during years 4 and 5, and yearly thereafter. DNA from peripheral blood mononuclear cells was also investigated from 9 stem cell donors before cell donation. Written informed consent was obtained according to the Declaration of Helsinki, and the study was approved by the institutional review board of Hannover Medical School (ethical vote 2179-2014).
Cytogenetic and molecular analyses
Preinduction chemotherapy blood or bone marrow samples were studied centrally by G- and R-banding analysis. Chromosomal abnormalities were described according to the International System for Human Cytogenetic Nomenclature.24 DNA was extracted by using the AllPrep DNA/RNA purification kit (Qiagen). DNA sequencing libraries were prepared from samples at diagnosis (N = 138) and at relapse (n = 47) with a custom TruSight myeloid sequencing panel according to the manufacturers’ instructions (Illumina), which included 46 entire genes or hotspots recurrently found in myeloid leukemias (supplemental Table 1). Host chimerism was examined by determining polymorphisms in short tandem repeats using polymerase chain reaction (PCR).
Error-corrected sequencing for sensitive MRD detection
We have developed an amplicon sequencing approach for sensitive detection of single nucleotide variants (SNVs) and insertions/deletions (indels) that significantly reduces the sequencing error rate, as previously described.14,25 In brief, we use a proofreading polymerase for PCR, random barcodes to allow bioinformatics error correction, perform the initial PCR with only 5 PCR cycles, and avoid identical multiplex identifier/gene combinations on consecutive MiSeq runs. The Illumina MiSeq reagent kit version 3 (600 cycles) was used for sequencing and was run on the MiSeq sequencer aiming for a high coverage per sample (we obtained, on average, 526 161 aligned reads per marker with 251 bases in both forward and reverse sequencing directions). This amplicon-based error-corrected sequencing and bioinformatics approach was applied to samples on day 90 (n = 133) and day 180 (n = 125) after alloHCT.
Bioinformatics and statistical analyses
Bioinformatics analysis of myeloid panel sequencing and of error-corrected sequencing was performed as previously described according to a standardized algorithm for calling SNVs and small and large indels MRD positive or negative based on the number of read families (RF mode, error-corrected sequencing) or the number of matching forward (R1) and reverse (R2) reads (R1/R2 mode), using the background error of the individual sample to define the limit of detection.14,25 Limit of detection for SNVs and small indels was defined as an average of the background error plus 3 standard deviations of the background error, in which background error is quantified by the largest non-reference variant allele fraction at all nucleotide positions between the primers of the respective amplicon. For large indels, ≥75 supporting (mutated) reads were required to call MRD positive, except for the NPM1 4 base pair insertion, in which the requirement was ≥10 supporting reads. If multiple gene mutations were used for MRD monitoring, the patient was defined as MRD positive when at least 1 of the mutations was called MRD positive.
Median follow-up time for survival was calculated with the reverse Kaplan-Meier method. Relapse was defined as blast increase to ≥5% in peripheral blood or bone marrow or evidence of extramedullary disease. OS end points, measured from the date of transplantation, were death (failure) and alive at last follow-up (censored). Relapse-free survival (RFS) end points, measured from the date of transplantation, were relapse (failure), death in complete remission (CR) (failure), and alive in CR at last follow-up (censored).1 The Kaplan-Meier method and log-rank tests were used to estimate the distribution of OS and RFS, and to compare differences between survival curves. The Gray test was used to compare and visually represent cumulative incidence of nonrelapse mortality (NRM) and CIR as competing risks using R package cmprsk.26
Categorized variables were considered in univariate analysis for CIR, NRM, RFS, and OS for the full data set and the non-DTA data set (supplemental Tables 7 and 10, respectively). Variables were used for multivariate analysis, if P was ≤.1 in univariate analysis and had distinct categorical values in at least 7 patients. For multivariate analysis, a Cox proportional hazards model was constructed for CIR, NRM, RFS, and OS adjusting for potential confounding covariates.27 Variables were reduced by backward elimination starting with all selected variables. At each step, the variable with the worst fit (judged from individual P values reported by the survival or the cmprsk packages of R) was removed from the model. The representative multivariate model was selected from this series of models, when the P values of all variables were smaller than .05.
Comparisons of variables were performed by using the Kolmogorov-Smirnov test and the χ2 test for categorical variables for exploratory purposes.
The prognostic impact of using one or multiple MRD markers was estimated by redefining MRD by either randomly selecting one gene as MRD marker or by omitting genes randomly 1000 times. From these analyses, the CIR was recalculated, and the distribution of the obtained hazard ratios is presented as histograms.
The two-sided level of significance was set at P < .05. The statistical analyses were performed with SPSS version 26.0 (IBM Corporation), statistical program R28 using packages “survival,” “cmprsk,” Microsoft excel 2010 (Microsoft Corporation), and custom linux scripts.
Results
Patients and MRD status after alloHCT
We included 154 patients in the genetic screening, of whom 138 (90%) had at least one molecular MRD marker and could be analyzed by using NGS-MRD. Patients with an MRD marker had a median age of 53 years (range, 19-74 years) and underwent alloHCT with pretransplant remission status of first or second CR/CR with incomplete hematologic recovery (n = 111 [80%]) or non-CR (n = 27 [20%]) between January 1998 and April 2017 (supplemental Table 2). MRD could be assessed before alloHCT in 81 of the CR/CR with incomplete hematologic recovery patients, and 42 were MRD positive (52%). In 120 patients (87%), MRD was assessed on days 90 and 180 after alloHCT and in 18 patients either on day 90 or day 180. A median of 2 molecular aberrations were used for MRD monitoring per patient (range, 1-5). In total, 495 MRD analyses were performed in 138 patients after alloHCT (472 in peripheral blood, 23 in bone marrow; 256 on day 90, 239 on day 180) (supplemental Table 3). Mutations in 42 different genes were used as MRD markers, frequently including DNMT3A, NPM1, IDH2, RUNX1, and FLT3 (supplemental Table 4; supplemental Figure 2). The median limit of detection was 0.0106% (range, 0.0016%-0.622%). The median variant allele frequency (VAF) of MRD-positive markers was 0.048% (range, 0.0034%-23.8%).
MRD and remission status before transplantation and on day 90 and 180 after transplantation are shown in supplemental Table 5. MRD was detected in 34 patients (25%) on day 90 and/or day 180 (29 [21%] on day 90, 18 [13%] on day 180). Clinical and transplantation-associated characteristics were similarly distributed between MRD-positive and MRD-negative patients except for a higher proportion of female donors in the MRD-positive group compared with the MRD-negative group (supplemental Table 2).
MRD status post-alloHCT significantly affects clinical outcome
The median follow-up time in our cohort was 6.0 years. Five-year CIR and OS for all patients were 33% and 68%, respectively. Twenty (59%) of 34 MRD-positive patients and 27 (26%) of 104 MRD-negative patients relapsed after alloHCT (total, n = 47 [34%]). By competing risk analysis for CIR and NRM, patients with positive MRD post-alloHCT had a 5-year CIR of 53% and MRD-negative patients of 26%, whereas NRM did not differ (CIR, P < .001; NRM, P = .198). In univariate analysis, RFS and OS were significantly shorter in MRD-positive patients compared with MRD-negative patients (supplemental Figure 3; supplemental Tables 6 and 7). In multivariate analysis, MRD positivity was an independent predictor of CIR and RFS, whereas it had no effect on NRM (P = .24) and OS (P = .07) (supplemental Table 6). Several well-known prognostic markers were not prognostic in our analysis, such as conditioning regimen, hematopoietic cell transplantation comorbidity index, and donor sex, whereas cytomegalovirus (CMV) status was, likely due to our patient selection criteria, which excluded patients with early relapse and death and required survival follow-up visits for at least 1 year.
DTA vs non-DTA mutations as MRD markers after alloHCT
Mutations in DNMT3A, TET2, and ASXL1 (DTA mutations) are associated with CH and were shown to be unsuitable markers of MRD for AML patients without or prior to alloHCT.13 We therefore evaluated the prognostic impact of DTA and non-DTA MRD positivity after alloHCT separately. In 51 patients, at least one DTA mutation was assessed post-alloHCT (supplemental Figure 4). Nine patients were MRD positive (17.6%; DNMT3A, n = 5, mean VAF = 0.024% [range, 0.009%-0.037%]; TET2, n = 3, mean VAF = 0.046% [range, 0.031%-0.047%]; ASXL1, n = 1, VAF on day 90 = 0.52%, VAF on day 180 = 23.8%), and 42 patients were MRD negative (82.3%). Five patients had at least one bone marrow sample assessed; all were MRD negative. Patient characteristics were similarly distributed between MRD-positive and MRD-negative patients with DTA mutations except for a higher proportion of female patients and of patients receiving RIC in the MRD-positive group (supplemental Table 8).
CIR, RFS, and OS were similar for MRD-positive and MRD-negative patients when DTA mutations were considered (Figure 1). To exclude donor origin of the DTA mutations, we sequenced peripheral blood mononuclear cells from the donors of the 9 patients with detectable MRD before stem cell harvest. In 2 donors, the same mutation as in the corresponding recipients’ diagnostic sample was detected at very low VAF (supplemental Figure 5A). We therefore repeated the prognostic evaluation in which the 2 patients with positive donors were considered MRD negative. Again, no prognostic effect of MRD in DTA genes was found for CIR, RFS, or OS (supplemental Figure 5B-D). In 3 patients, MRD increased from day 90 to day 180, which correlated well with relapse: 2 of these patients developed a relapse at days 210 and 272, respectively, and the third patient had a decreasing chimerism and received prophylactic donor lymphocyte infusions on day 344. This reestablished full chimerism, and the patient is alive 6 years after transplantation without relapse. Of the remaining 6 DTA-MRD–positive patients, only 1 patient relapsed (at day 691).
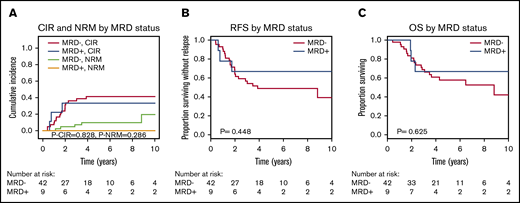
CIR, NRM, RFS, and OS for patients who were MRD positive on day 90 or 180 compared with MRD-negative patients according to mutations in DTA genes (51 patients). (A) CIR and NRM by competing risk analysis for MRD-positive (n = 9) and MRD-negative (n = 42) patients. (B) RFS for MRD-positive (n = 9) and MRD-negative (n = 42) patients. (C) OS for MRD-positive (n = 9) and MRD-negative (n = 42) patients.
CIR, NRM, RFS, and OS for patients who were MRD positive on day 90 or 180 compared with MRD-negative patients according to mutations in DTA genes (51 patients). (A) CIR and NRM by competing risk analysis for MRD-positive (n = 9) and MRD-negative (n = 42) patients. (B) RFS for MRD-positive (n = 9) and MRD-negative (n = 42) patients. (C) OS for MRD-positive (n = 9) and MRD-negative (n = 42) patients.
In a separate analysis, only non-DTA mutations were considered in a group of 131 patients (supplemental Figure 4). Twenty-six patients in the non-DTA group were MRD positive (19.8%) compared with 105 patients who were MRD negative (80.2%). Ten patients had at least one bone marrow sample assessed, of which two were MRD positive (MRD rate in bone marrow, 20% [ie, similar to peripheral blood]). Patient characteristics were similarly distributed between MRD-positive and MRD-negative patients (Table 1). Molecular mutations occurring in ≥6 patients at time of diagnosis and the mutation classes did not correlate with MRD after alloHCT (supplemental Table 9).
Interestingly, non-DTA mutation–based MRD was highly predictive for CIR, RFS, and OS in univariate and multivariate analyses (Figure 2; Table 2; supplemental Table 10). Subgroup analysis showed that the prognostic effect of non-DTA MRD on CIR was consistent across most clinical and genetic subgroups, including European LeukemiaNet risk groups (Figure 3). NGS-MRD and conventional donor chimerism analysis resulted in a similarly high specificity (0.91 and 0.89) and moderate sensitivity (0.40 and 0.36), whereas the combination of both analyses increased sensitivity and maintained specificity (0.51 and 0.83), respectively (supplemental Table 11).

CIR, NRM, RFS, and OS for patients who were MRD positive on day 90 or 180 compared with MRD-negative patients according to mutations in non-DTA genes (131 patients). (A) CIR and NRM by competing risk analysis for MRD-positive (n = 26) and MRD-negative (n = 105) patients. (B) RFS for MRD-positive (n = 26) and MRD-negative (n = 105) patients. (C) OS for MRD-positive (n = 26) and MRD-negative (n = 105) patients.
CIR, NRM, RFS, and OS for patients who were MRD positive on day 90 or 180 compared with MRD-negative patients according to mutations in non-DTA genes (131 patients). (A) CIR and NRM by competing risk analysis for MRD-positive (n = 26) and MRD-negative (n = 105) patients. (B) RFS for MRD-positive (n = 26) and MRD-negative (n = 105) patients. (C) OS for MRD-positive (n = 26) and MRD-negative (n = 105) patients.

Forest plot showing the prognostic impact of MRD positivity on CIR in patient subsets. aGvHD, acute-graft-versus-host disease; cGvHD, chronic graft versus host disease; ELN, European LeukemiaNet; FLT3-ITD, FLT3–internal tandem duplication; HCT-CI, hematopoietic cell transplantation comorbidity index.
Forest plot showing the prognostic impact of MRD positivity on CIR in patient subsets. aGvHD, acute-graft-versus-host disease; cGvHD, chronic graft versus host disease; ELN, European LeukemiaNet; FLT3-ITD, FLT3–internal tandem duplication; HCT-CI, hematopoietic cell transplantation comorbidity index.
In relapsing patients, the median time to relapse was similar between MRD-positive and MRD-negative patients (P = .209) (supplemental Figure 6), and the VAF of MRD-positive patients did not correlate with time to relapse (data not shown). Eighteen patients were MRD positive and relapsed; in those, MRD was found in 17 non-DTA genes, indicating that a wide variety of genes contributed to MRD positivity (supplemental Table 12). A CMV serostatus other than donor and patient negativity was associated with significantly higher CIR and shorter RFS and OS. CMV reactivation was strongly associated with CMV serostatus and was also associated with higher CIR and shorter RFS and OS (supplemental Figure 7). However, in multivariate analysis, only CMV serostatus remained significant in the non-DTA cohort.
Log-reduction, MRD cutoff, and pretransplant characteristics
We next evaluated whether a 3-log reduction of MRD between diagnosis and post-alloHCT MRD time points was a prognostic MRD cutoff in patients with mutations in non-DTA genes. MRD-positive patients with less or more than a 3-log MRD reduction had a similarly increased CIR and decreased RFS and OS compared with MRD-negative patients, suggesting that MRD positivity after transplantation is prognostic independent of the level of log-reduction (supplemental Figure 8).
The impact of the VAF on relapse risk was also evaluated for the VAF cutoffs 0.1% and 1% based on non-DTA mutations. Twelve patients had at least one MRD marker with a VAF >0.1% and 5 patients with a VAF >1%. CIR, RFS, and OS were similar among MRD-positive patients independent of the VAF cutoff. In supplemental Figures 9 and 10, panels A, B, and C are based on the MRD marker with the highest VAF if multiple markers were measured; panels D, E, and F are based on the MRD marker with the lowest VAF if multiple markers were measured.
We also evaluated the role of post-alloHCT MRD depending on pre-alloHCT MRD status in patients who were in CR before alloHCT. Data were available for 77 patients when restricting the analysis to mutations in non-DTA genes. Patients who were MRD negative before alloHCT had an excellent prognosis independent of post-alloHCT MRD. Patients who were MRD positive before alloHCT and who became negative after alloHCT had an intermediate prognosis, whereas patients who were MRD positive before and after alloHCT had the worst prognosis (supplemental Figure 11).
Evaluating conditioning intensity in CR patients who were either MRD positive or negative before alloHCT based on non-DTA mutations, we found no prognostic effect of conditioning intensity (MAC or RIC) for CIR, RFS, or OS (supplemental Figure 12).
We then systematically evaluated the impact of using 1, 2, or all minus 1 of the available MRD markers per patient for MRD monitoring. MRD markers were selected randomly by 1000 permutations, and the resulting hazard ratios for CIR of MRD-positive patients are plotted in supplemental Figure 13. Using >1 MRD marker per patient decreased the width of the hazard ratio distribution (thus making it better defined) and tended to increase the hazard ratios, underscoring the benefit of using multiple MRD markers for each patient.
Discussion
Evaluating MRD status has gained increasing interest in AML therapy with the advancement of novel techniques.29-33 Importantly, MRD negativity is emerging as an alternative therapeutic end point that could supplement or replace standard AML CR critieria.23 Although numerous studies have shown the utility of MRD outside alloHCT13,34,35 or before alloHCT,14,15,30,36-40 MRD is not well studied after alloHCT. Here, we describe a molecular MRD approach after alloHCT that is based on the mutational profile at the time of diagnosis, uses error-corrected sequencing that reaches sensitivity levels as low as 10−4, and uses multiple MRD markers per patient. Our study found that NGS-based MRD monitoring of non-DTA mutations on day 90 and 180 after alloHCT is independently predictive for CIR, RFS, and OS in patients with AML. The prognostic impact was independent of the degree of MRD positivity (VAF) or the level of log reduction (less or more than a 3-log MRD reduction).
Molecular MRD is challenged by mutations that derive from preleukemic clones.23 After alloHCT, the recipient’s hematopoiesis is replaced by the donor’s so that CH-type genetic abnormalities should be eliminated following successful alloHCT. It has therefore been speculated that CH-type mutations are reliable MRD markers post-alloHCT, as they indicate persistence or relapse of recipient hematopoiesis and the leukemic clone.10 In the current study, DTA mutations were not eliminated in 17.6% of patients with DTA mutations. DTA mutations were mostly present at very low VAF after alloHCT and may represent residual CH of the recipient. In 2 of 9 patients, the same DTA mutation of the patient was also found in donor cells at a time before stem cell harvest, and therefore the origin of the mutated clone cannot be resolved. However, even if the patients with potential donor origin of MRD were considered MRD negative, MRD in DTA mutations had no prognostic effect in our study when a single time point was analyzed. In contrast, an increasing VAF from day 90 to day 180 correlated with relapse or declining chimerism, suggesting that the kinetics of DTA mutations may be a better indicator of potential relapse. This was similarly observed by Brambati et al,41 who evaluated MRD in 17 patients post-alloHCT using DNMT3A und IDH1/2 mutations. Although MRD was detectable shortly before relapse, none of the 5 patients who relapsed after day 90 were MRD positive on day 90. Although non-DTA mutations are clearly prognostic after alloHCT, the prognostic value of DTA mutations post-alloHCT requires further validation in additional patient cohorts.
Few studies have thus far analyzed MRD status after alloHCT using NGS42 or multiparametric flow cytometry.43,44 Using multiparametric flow cytometry for MRD assessment, only 3% to 6% of patients were found to be MRD positive after 1 to 6 months’ post-alloHCT compared with 23% to 31% prior to alloHCT. Kim et al42 applied a NGS-based MRD approach on day 21 after alloHCT and found 15.4% of patients to be MRD positive with a VAF cutoff ≥0.2%. In our study, 25% of patients were MRD positive when a low VAF cutoff was applied (median, 0.0106%) and when MRD was assessed at later time points (days 90 and 180 post-alloHCT). Although early detection of MRD is desirable, it should be kept in mind that MRD assessment as early as 1 month after alloHCT may be confounded by residual recipient hematopoiesis, especially after RIC, as the patient’s hematopoiesis is only gradually replaced by that of the donor’s.45 In line with this, 11 patients in our cohort were MRD positive on day 90 but negative on day 180, of whom 7 were conditioned with reduced intensity (none received additional treatment between day 90 and day 180), suggesting an ongoing graft vs leukemia effect during this period.
We found that almost all patients who were MRD positive before alloHCT and who remained MRD positive on day 90 post-alloHCT relapsed with a 5-year CIR of 90%. This finding suggests that MRD positivity prior to and post-alloHCT is the strongest predictor for relapse and that MRD status prior to and post-alloHCT should be considered complementary.
Hourigan et al15 showed that MRD positivity before alloHCT only affected CIR and OS in patients receiving RIC but not in patients receiving MAC, suggesting that MRD-positive patients benefit from MAC. Similarly, lower conditioning intensity was associated with a higher relapse incidence only in MRD-positive but not MRD-negative patients in a large registry analysis.46 In contrast, a large meta-analysis of mostly multiparametric flow cytometry MRD studies concluded that MAC was not able to attenuate the negative prognostic impact of pre-alloHCT MRD positivity.47 In our study, conditioning intensity was not associated with posttransplant relapse risk and RFS in patients who were in CR before alloHCT.
Our results are primarily based on peripheral blood samples (472 analyses on peripheral blood vs 23 on bone marrow), which encourage the use of peripheral blood as a source for MRD monitoring post-alloHCT and reduce the burden of routine bone marrow biopsies. In the future, it should be evaluated whether analysis of bone marrow post-alloHCT may increase the proportion of MRD-positive patients and may improve risk stratification.
We found that the use of multiple MRD markers per patient improves the diagnostic accuracy. Ten percent of the relapsing patients did not preserve any diagnostic mutation at relapse and therefore may be missed by MRD monitoring that relies on diagnostic mutations; future efforts should therefore aim at using all available known mutations for MRD monitoring and at improving sensitivity of panel approaches that also allow detection of de novo mutations in CR samples.
Our study has several limitations. It is a retrospective study, includes patients who were conditioned for alloHCT with regimens of different intensity (RIC and MAC), and can not separately assess the role of MRD assessment in peripheral blood vs bone marrow samples due to the limited number of bone marrow samples available. In addition, it is not fully representative of an average AML transplant cohort due to our selection criteria in which relapse before day 90 was excluded and survival follow-up of at least 1 year was mandatory. Nevertheless, our selection criteria assure that the cohort is sufficiently homogeneous to allow the prognostic comparison between MRD-positive and MRD-negative patients.
In conclusion, our study showed that NGS-MRD after alloHCT is prognostic when non-DTA mutations are used, is feasible from peripheral blood samples, and is most predictive in patients who are MRD positive before alloHCT.
This work was presented in part at the 2019 annual meeting of the American Society of Hematology, Orlando, FL, 7-10 December 2019.
All sequencing results are provided in supplementary materials. Flow cytometry standard files from measurable residual disease analyses can be requested from the corresponding authors. The full sequencing data cannot be made available, to comply with the EU General Data Protection Regulation.
Acknowledgments
The authors thank all participating patients and contributing nurses and doctors, as well as Kerstin Görlich, Patricia Hanel, Silvia Horter, Monika Krappe, Marlene Reuter, and Anja Ziolek for their support.
This work was supported by grant DJCLS 06 R/2017 from Deutsche José Carreras Stiftung, grants 109714 and 70112697 from Deutsche Krebshilfe, grant 01GM1909A from Bundesministerium für Bildung und Forschung, an European Research Council grant under the European Union’s Horizon 2020 research and innovation program (No. 638035), and German Research Foundation (DFG) grants HE 5240/5-1, HE 5240/5-2, HE 5240/6-1, and HE 5240/6-2.
Authorship
Contribution: M.H. and F.T. designed the study; M.H., B.H., K.B., C.P.W., K.T., C.F., M.B., P.K., A.L., M.W., B.N., A.C., A.K., K.M., L.H., R.G., and F.T. performed the research; M.H., V.I.G., P.P., L.B., W.F., A.H., W.P., J.K., K.D., H.D., A.G., M.S., L.H., and F.T. provided study materials; M.H. and F.T. wrote the manuscript; and all authors analyzed and interpreted the data, and agreed to the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Felicitas Thol, Department of Hematology, Hemostasis, Oncology and Stem Cell Transplantation, Hannover Medical School, Carl-Neuberg Str 1, 30625 Hannover, Germany; e-mail: thol.felicitas@mh-hannover.de; or Michael Heuser, Department of Hematology, Hemostasis, Oncology and Stem Cell Transplantation, Hannover Medical School, Carl-Neuberg Str 1, 30625 Hannover, Germany; e-mail: heuser.michael@mh-hannover.de.



