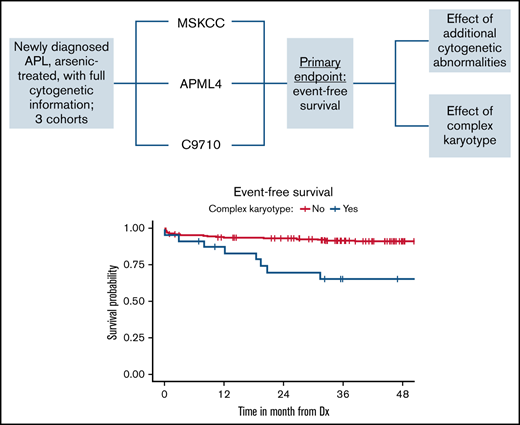
Key Point
Complex karyotype is associated with inferior event-free survival in patients who have acute promyelocytic leukemia treated with ATO.

Abstract
Frontline arsenic trioxide (ATO)–based treatment regimens achieve high rates of long-term relapse-free survival in treating acute promyelocytic leukemia (APL) and form the current standard of care. Refining prognostic estimates for newly diagnosed patients treated with ATO-containing regimens remains important in continuing to improve outcomes and identify patients who achieve suboptimal outcomes. We performed a pooled analysis of exclusively ATO-treated patients at a single academic institution and from the ALLG APML4 and Alliance C9710 studies to determine the prognostic importance of additional cytogenetic abnormalities and/or complex karyotype. We demonstrated inferior event-free survival for patients harboring complex karyotype (hazard ratio [HR], 3.74; 95% confidence interval [CI], 1.63-8.56; P = .002), but not for patients harboring additional cytogenetic abnormalities (HR, 2.13; 95% CI, 0.78-5.82; P = .142). These data support a role for full karyotypic analysis of all patients with APL and indicate a need for novel treatment strategies to overcome the adverse effect of APL harboring complex karyotype.
Introduction
Contemporary risk stratification of patients with acute promyelocytic leukemia (APL) is based solely on the degree of leukocytosis at diagnosis, and this delineation influences therapeutic decision making in clinical practice.1 Although many prognostic variables have been investigated to stratify patients, the data are conflicting concerning the prognostic relevance of additional cytogenetic abnormalities (ACAs) that may be present beyond t(15;17) and of complex karyotype (CK) and whether the presence of such abnormalities should influence decisions for treating patients with APL.2-8 Prior efforts have been limited by heterogeneous cohorts and small samples of patients.
This issue is especially uncertain in patients who receive arsenic trioxide (ATO)–containing frontline treatment regimens, because most prior studies investigating cytogenetic abnormalities and clinical outcomes in APL either included patients treated without ATO or analyzed ATO- and non-ATO–treated patients together. Furthermore, delineating the potential prognostic importance of ACA and CK is particularly timely, given that ATO-containing regimens are the current standard of care for all patients with newly diagnosed APL.1,9 Therefore, we sought to determine the impact of ACA and CK on event-free survival (EFS) in ATO-treated patients with APL, hypothesizing that the presence of ACA or CK may portend inferior clinical outcomes.
Materials and methods
We studied all patients with APL evaluated at Memorial Sloan Kettering Cancer Center (MSKCC) since 2005. Only patients who began induction therapy with an ATO-based regimen that included all-trans retinoic acid (ATRA) were included in this analysis, to ensure uniformity of the study population and applicability of results to contemporary clinical practice in APL. We also included patients treated in the Australasian Leukaemia and Lymphoma Group APML4 study10 (frontline ATRA plus ATO plus idarubicin), and the ATO-treated subset of patients in the Alliance C9710 study11 (ATRA plus chemotherapy induction; sequential ATO followed by ATRA/chemotherapy consolidation; and finally, intermittent ATRA maintenance therapy vs observation). The protocol regimens and logistics are detailed in the descriptions of each trial.10,11 Patients in the APML4 study underwent polymerase chain reaction (PCR)-based molecular surveillance on bone marrow samples while in remission, every 3 months for 36 months, which corresponds to 12 months after completion of maintenance therapy. In the C9710 study, paired bone marrow aspirate/biopsy and peripheral blood PCR testing were performed after documentation of complete response (CR) or partial response after induction therapy. In addition, patients in the ATO arm underwent testing after the last cycle of ATO; after cycles 1 and 2 of ATRA/daunorubicin consolidation; at 4, 8, and 12 months during maintenance; every 6 months for 1 year; yearly for 2 years; and as needed thereafter, based on clinical concern (eg, alterations in peripheral blood counts suggesting relapse) or to confirm overt relapse. Of note, previous publications have examined the influence of CK within the C97105,7 and APML410 studies separately. This study was approved by the institutional review board at MSKCC and was conducted in accordance with the Declaration of Helsinki.
We included all patients with a baseline, pretreatment karyotype demonstrating the presence of t(15;17) and 1 patient without karyotype but with fluorescence in situ hybridization (FISH) at diagnosis that revealed the presence of an ACA. We excluded patients with PML-RARA solely, detected by FISH or PCR assays. CK status was available for the entire C9710 data set of 93 patients and ACA status for a subset therein of 33 patients. Manual chart review extracted patient, disease, and clinical data for patients treated at MSKCC. We excluded patients without clinical follow-up information and patients who relapsed before transferring care to MSKCC. In addition, we excluded patients with APL harboring non-PML-RARA cytogenetic variants, given their potential influence on outcome.
We defined ACA as the presence of ≥1 cytogenetic abnormality besides t(15;17) and CK as the presence of ≥2 ACAs in addition to t(15;17). We defined coagulopathy as activated partial thromboplastin time/mean laboratory normal activated partial thromboplastin time >1.5, international normalized ratio >1.5 (or prothrombin time/mean laboratory normal prothrombin time >1.5 when the International Normalized Ratio and International Sensitivity Index were unavailable), or fibrinogen <100 mg/dL. The primary end point was EFS, which we defined as the time from diagnosis (MSKCC) or trial registration (APML4 and C9710) to either APL relapse or death, and the patients alive at last follow-up were censored. We assessed associations between time-to-event outcomes and patient and disease characteristics that potentially influenced outcomes: patient sex, age (>60 years and 45-60 years each vs <45 years), risk categorization according to presenting white blood cell (WBC) count (≥10 vs. <10, 103/μL), and presence of coagulopathy, ACA, and CK, in univariate Cox proportional hazards models. Fixed-effect meta-analysis methods were used to combine estimates from the 3 data sets queried.
Results
A total of 248 patients were included (MSKCC; n = 47; APML4, n = 108; C9710, n = 93); 6 patients from the MSKCC cohort were excluded because of APL relapse before the initial visit and 1 from APML4 because of the lack of follow-up information. Table 1 displays demographic information, baseline clinical and disease characteristics, and treatments received. The mean age at diagnosis was 47 years in the MSKCC cohort, 43 years in the APML4 cohort, and 42 years in the C9710 cohort. We excluded coagulopathy data from the C9710 cohort in our analyses, given a higher-than-expected rate of hypofibrinogenemia that classified all patients as having coagulopathy. Median follow-up among surviving patients was 36 months for MSKCC (range, 2-144), 54 months for APML4 (range, 28-96), and 98 months for C9710 (range, 95.5-105.1).
In total, 248 patients were analyzed: 188 cases had detailed information regarding ACA treatment available, and 247 had information regarding CK status. Fifty-nine of 188 (31%) patients had ACAs, and 25 (10%) harbored CK (13% MSKCC; 10% APML4: denominator excludes 1 patient with single ACA by FISH; and 9% C9710). The most common ACA was trisomy 8, detected in 30 patients (51% of patients with ACA), followed by deletion of 9q in 7 patients (12%); 4 other abnormalities were each seen in 2 patients, and all others were observed in only 1 patient in the MSKCC and APML4 cohorts (Table 1).
Survival for all 3 cohorts is displayed in Figure 1 for patients with available data and demonstrate inferior outcomes for MSKCC and C9710 (EFS/overall survival) in nonlandmark analyses (Figure 1A-B) and equivalent outcomes in 2-month landmark analyses (Figure 1C-D). Examining the influence of ACAs and CK on survival outcomes in pooled analyses (Figures 2 and 3) showed that EFS did not differ according to ACA status (univariate HR, 2.13; 95% CI, 0.78-5.82; P = .142), but that the presence of CK conferred inferior EFS (univariate HR, 3.74; 95% CI, 1.63-8.56; P = .005). These associations held true in multivariable analyses (Figure 3C-D). A high-risk disease state (elevated WBC count) and older age (>60 vs <45 years) also conferred inferior EFS in these analyses. We performed a 2-month landmark analysis to censor patients who died early in the disease course and to solely examine the effect on relapse. In these analyses, CK (HR, 5.1; 95% CI, 1.98-13.16; P = .001) and high-risk WBC (HR, 2.5; 95% CI, 1.01-6.17; P = .047) maintained influence, whereas older age (HR: 2.83, 95% CI: 0.82-9.8, P = .101) no longer met the threshold for statistical significance. (Results of these analyses conducted separately for each cohort are included in the supplement.)
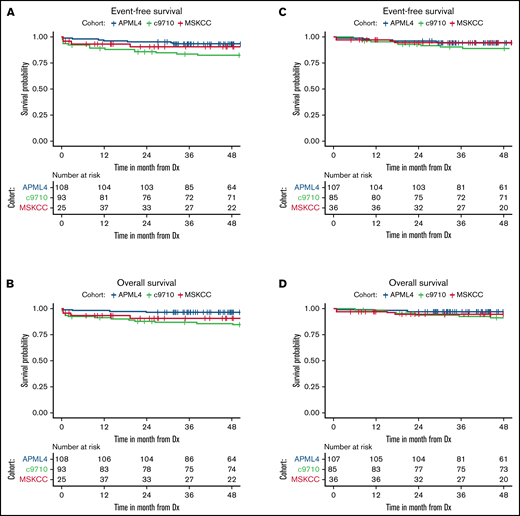
Patient survival in non-landmark and 2-month landmark analyses according to cohort. Event-free (A) and overall (B) survival of patients from time of diagnosis in non-landmark analyses. Event-free (C) and overall (D) survival of patients in landmark analyses from 2 months after diagnosis. Dx, diagnosis.
Patient survival in non-landmark and 2-month landmark analyses according to cohort. Event-free (A) and overall (B) survival of patients from time of diagnosis in non-landmark analyses. Event-free (C) and overall (D) survival of patients in landmark analyses from 2 months after diagnosis. Dx, diagnosis.
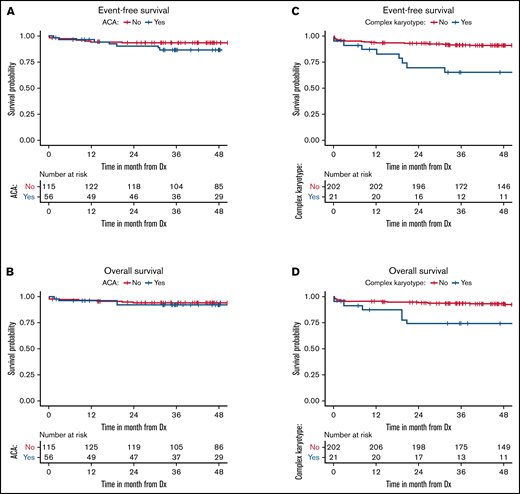
Patient survival according to ACA and CK status. Event-free (A) and overall (B) survival of patients according to ACA status. Event-free (C) and overall (D) survival of patients according to CK status. Dx, diagnosis.
Patient survival according to ACA and CK status. Event-free (A) and overall (B) survival of patients according to ACA status. Event-free (C) and overall (D) survival of patients according to CK status. Dx, diagnosis.
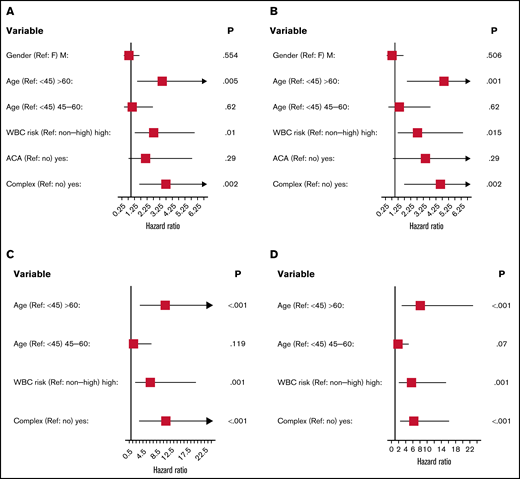
Forrest plots of prognostic factors for event-free and overall survival in univariate and multivariable analyses. Event-free/univariate (A), overall/univariate (B), event-free/multivariable (C), and overall survival/multivariable (D). High-risk WBC is >10 (103/μL) at diagnosis. ACA indicates additional chromosomal abnormalities; CK was defined as 2 or more chromosomal abnormalities in addition to t(15;17).
Forrest plots of prognostic factors for event-free and overall survival in univariate and multivariable analyses. Event-free/univariate (A), overall/univariate (B), event-free/multivariable (C), and overall survival/multivariable (D). High-risk WBC is >10 (103/μL) at diagnosis. ACA indicates additional chromosomal abnormalities; CK was defined as 2 or more chromosomal abnormalities in addition to t(15;17).
Five patients in the APML4 study relapsed, including 2 with CK and 3 without CK. Among patients with CK, 1 experienced molecular relapse at day 362 after achieving CR and was reinduced with ATRA+ATO followed by high-dose therapy with autologous stem cell rescue; the other patient with CK experienced molecular relapse at day 784 after CR without subsequent clinical follow-up information. Among patients without CK, 1 experienced bone marrow molecular relapse with overt central nervous system involvement at day 168 after CR. This patient underwent reinduction and consolidation with high-dose therapy with autologous stem cell rescue, but ultimately died of progressive APL. Another patient also experienced molecular relapse at day 189 after CR and succumbed to infection after salvage chemotherapy. The final patient harbored an ACA and experienced hematological relapse at day 742 after CR, but lacked subsequent clinical follow-up information.
Discussion
We present, to our knowledge, the largest published cohort containing exclusively ATO-treated patients with APL with detailed cytogenetic characterization to determine the prognostic effect of ACA and CK. In this pooled analysis from a large single-center experience and 2 prospective, cooperative group trials, the presence of an ACA beyond t(15;17) did not influence EFS. However, our data demonstrate that CK negatively influenced EFS. Censoring for early death (within 2 months of diagnosis) supported these same findings. These results contribute to our understanding of APL risk stratification and should inform future clinical studies of this disease.
Other groups have examined the effect of karyotypic abnormalities on survival in APL. In ATRA-treated patients, de Botton and colleagues8 showed no influence of ACA on cumulative incidence of relapse, EFS, and overall survival, but CK was not included in their analysis. More recently, the Spanish PETHEMA group2 analyzed >1500 patients with APL and found an increased risk of relapse in patients with CK treated with ATRA plus chemotherapy. A large series examining prognostic factors in APL with exclusively ATO-treated patients was published by Lou et al3 including 184 patients, but no analyses were conducted to examine the influence of ACA or CK on survival outcomes.
Clinical data using single-agent ATO (that is, without ATRA or chemotherapy) show favorable efficacy and safety.12,13 In this context, Mathews and colleagues14 examined the prognostic influence of ACA in 69 patients (23% harboring ACA) treated with single-agent ATO at a single institution. There were no differences in baseline clinical characteristics between their patients who had ACAs and those who did not. Importantly, they did not detect differences in time until hematologic/molecular remission or survival outcomes between the 2 groups; the effect of CK was not examined in their analyses. The published experience using single-agent ATO12 demonstrated a steady trend of relapses that plateaued at ∼2 years after diagnosis in this context, with most relapses occurring within 1 year.
One noteworthy difference between the C9710 and APML4 trials was the timing of ATO administration: in C9710, ATO was not given during induction, whereas in APML4 the ATO was administered earlier in the course of treatment. Although our analysis is limited by small sample groups, it may suggest that earlier treatment with ATO could overcome negative prognostic factors, such as the CK that was observed in C9710.
Besides demonstrating the clinical impact of CK on EFS in patients with APL, our results reinforce the importance of pursuing full karyotypic analysis rather than relying solely on PCR- or FISH-based testing of patients with suspected or confirmed APL. This recommendation is endorsed by consensus peer-reviewed, evidence-based guidelines for the diagnostic evaluation of acute leukemia.9,15 Although PCR- and FISH-based testing offer more rapid results in urgent clinical situations and require less intensive technician time and effort than conventional karyotype analysis, the added prognostic information of CK status (approximately half of patients with ACAs) appears valuable and may serve to garner further data to study this question.
There are limitations to our study. First, although all patients received ATO for frontline treatment, the additional components of treatment varied between patients and across settings in our meta-analysis and outcomes varied across the studies (Figure 2), introducing heterogeneity. Second, with limited data from cooperative group studies and in retrospective analyses, additional clinicopathologic parameters that may influence clinical outcomes, such as FMS-related tyrosine kinase 3 (FLT3) or TP53 mutational status,10,16 which may covary with the presence of CK, could not be included in our study across all patients in these analyses. Future clinical studies may benefit from incorporating more widespread mutational profiling to further refine our ability to estimate the prognosis for patients with APL, as is routine standard of care for patients with non-APL acute myeloid leukemia.9 The lack of clinical impact of FLT3 mutation in APL was shown in the C97105 and APML410 trials and in the context of single-agent ATO.14 Finally, although we sought to assemble a large cohort of uniformly treated patients, we recognize that our final sample size of 248 patients still limited our statistical power in potentially detecting moderate effects for the prognostic variables that we examined.
In summary, in a large, pooled population of exclusively ATO-treated patients with APL, CK, but not ACA, carried prognostic importance and conferred inferior EFS. In future studies, additional data should be collated from prospective trials to garner greater power to further address the question of the influence of CK on EFS and to confirm these preliminary findings. If these results are validated, treatment paradigms could be altered. For example, intensifying (eg, addition of cytotoxic chemotherapy when it otherwise would not be used) or prolonging (eg, extending consolidation duration) different phases of treatment could be investigated in prospective studies toward overcoming this adverse effect and improving outcomes for patients with APL who harbor CK.
Acknowledgments
The authors thank Janey Stone and Andrew Wei (Australasian Leukaemia and Lymphoma Group), Joseph Schmeltz (MSKCC), and Anne Arezina (Alliance for Clinical Trials in Oncology) for assistance with data collection.
This work was supported by an institutional grant from the National Institutes of Health, National Cancer Institute (NCI) grant P30CA008748; an AACR-AstraZeneca Lymphoma Research Fellowship and support through the Lymphoma Research Foundation’s Lymphoma Scientific Research Mentorship Program and the AIDS Malignancy Consortium’s Domestic Scholar Program (Z.D.E-P); and an institutional cancer center grant from the NCI P30 CA008748 (Z.D.E.P., A.D., Y.Z., and M.S.T.). Support for the C9710 trial was provided by a grant from the NCI to Cancer and Leukemia Group B (CALGB; CA31946; Durham, NC) and by NCI grant CA33601 to the CALGB Statistical Center.
Authorship
Contribution: Z.D.E-P and M.S.T. conceived of and designed the study; A.D. conducted biostatistical analyses and created figures; S.G., K.M., J.K., H.I., L.J.C., R.A.L., X.P., B.L.P., W.S., R.M.S., and M.S.T. designed and conducted the cooperative group clinical trials included hereinand provided data for analysis; J.H.P., S.R., and E.M.S. assisted with study conception and provided critical suggestions and feedback; Y.Z. provided input and oversight into cytogenetic analyses; and all authors read and approved the manuscript.
Conflict-of-interest disclosure: M.S.T. declares research funding from Orsenix. M.S.T. and R.M.S. serve on the advisory board for Syros Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Zachary Epstein-Peterson, Memorial Sloan Kettering Cancer Center, 530 East 74th St, New York, NY 10021; e-mail: epsteinz@mskcc.org.