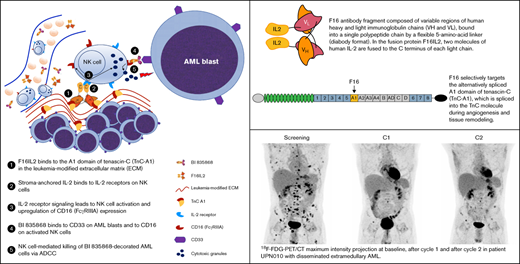
Key Points
F16-monoclonal antibody–mediated delivery of IL-2 to the leukemia-modified stroma induced an expansion and activation of NK cells.
F16IL2/BI 836858 combination therapy showed a manageable safety profile and resulted in clinical responses in posttransplant AML relapse.

Abstract
Natural killer (NK) cells are key effectors in cancer immunosurveillance and posttransplant immunity, but deficiency of environmental signals and insufficient tumor recognition may limit their activity. We hypothesized that the antibody-mediated anchoring of interleukin-2 (IL-2) to a spliced isoform of the extracellular matrix (ECM) glycoprotein tenascin-C would potentiate NK-cell–mediated antibody-dependent cellular cytotoxicity against leukemic blasts. In this novel-novel combination, dose-escalation, phase 1 trial, we enrolled patients with posttransplant acute myeloid leukemia (AML) relapse to evaluate the safety, pharmacokinetics, pharmacodynamics, and preliminary activity of the antibody-cytokine fusion F16IL2 (10 × 106 to 20 × 106 IU IV; days 1, 8, 15, and 22 of each 28-day cycle) in combination with the anti-CD33 antibody BI 836858 (10-40 mg IV, 2 days after each F16IL2 infusion). Among the 15 patients (median [range] age, 50 [20-68] years) treated across 4 dose levels (DLs), 6 (40%) had received 2 or 3 prior transplantations. The most frequent adverse events were pyrexia, chills, and infusion-related reactions, which were manageable, transient and of grade ≤2. One dose-limiting toxicity occurred at each of DLs 3 (pulmonary edema) and 4 (graft-versus-host disease). Three objective responses were observed among 7 patients treated at the 2 higher DLs, whereas no responses occurred at the 2 starting DLs. Combination therapy stimulated the expansion and activation of NK cells, including those expressing the FcγRIIIA/CD16 receptor. ECM-targeted IL-2 combined with anti-CD33 immunotherapy represents an innovative approach associated with acceptable safety and encouraging biologic and clinical activity in posttransplant AML relapse. This trial was registered at EudraCT as 2015-004763-37.
Introduction
Patients with acute myeloid leukemia (AML) who relapse after allogeneic hematopoietic stem cell transplantation (HSCT) have a dismal prognosis.1,2 Available therapeutic options include donor lymphocyte infusions, hypomethylating agents alone or in combination, chemotherapy, and a second allograft, but long-term benefit is usually limited to those who are eligible for a second immune cell-based intervention.1,3-5
Natural killer (NK) cells eliminate cancer cells through the release of cytotoxic granules triggered by interactions with natural ligands or through FcγRIIIA/CD16-mediated recognition of antibody-decorated cells, in a process called antibody-dependent cellular cytotoxicity (ADCC).6 The cytokines of the common γ-chain family interleukin-2 (IL-2) and IL-15 endow NK cells with improved effector functions and extend their persistence in vivo.6-8 Shortage of cytokines is a crucial factor that limits NK cell activity in the tumor microenvironment.6 However, attempts to administer therapeutically relevant doses of unmodified cytokines are hindered by unfavorable pharmacokinetic properties and substantial side effects.9 The multikinase inhibitor sorafenib has recently been shown as a means to increase IL-15 levels locally in the leukemic bone marrow (BM) in posttransplant FLT3-mutant AML by enhancing IL-15 gene transcription in leukemic cells.10 A more versatile avenue to improve the therapeutic index of cytokines is to use mAbs as delivery vehicles to localize cytokine effects directly to the tumor microenvironment.11-13 In this context, attaching cytokines to extracellular matrix (ECM) rather than cell surface antigens is particularly attractive, given the high abundance and stability of target expression in the stroma, the intrinsic accessibility from the bloodstream and the lack of constitutive depletion of cytokine fusions as seen in the steady-state internalization of membrane receptors.12-14
F16IL2 is an investigational fully human recombinant fusion protein composed of the antibody fragment scFv(F16) in diabody format and 2 molecules of IL-2 fused to the C terminus of each light immunoglobulin chain (Figure 1A).15,16 F16 specifically targets the A1 domain of tenascin-C (TnC), which is spliced into TnC during angiogenesis and tissue remodeling (Figure 1B). TnC-A1 is expressed in the subendothelial ECM of solid and hematological malignancies, but is virtually absent in healthy organs, enabling a clear-cut discrimination between normal and diseased tissues.12,13,16 The F16IL2 fusion mediates the site-specific delivery of IL-2 to and its physical retainment at the tumor site, thereby inducing tumor regression in various models of solid cancer.15,17 We have shown that IL-2-based immunocytokines are also highly active against hematological malignancies, including AML, which abundantly express TnC-A1.18-20 A murine analog of F16IL2 induced a strong inhibition of AML progression in immunocompromised (but NK cell–competent) and immunocompetent mice, whereas equivalent doses of nontargeted IL-2 were ineffective.20 When combined with cytarabine, durable leukemia remissions were achieved. The therapeutic efficacy was dependent on NK and CD8+ T cells, as depletion of either of these subsets completely abrogated therapeutic activity.20
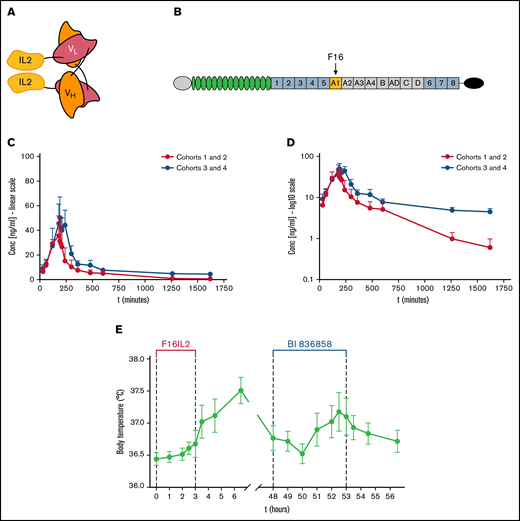
The F16IL2 immunocytokine and its ECM target TnC-A1, F16IL2 pharmacokinetics, and body temperature changes after F16IL2 and BI 836858 administration. (A) The F16 antibody fragment is composed of variable regions of human heavy and light immunoglobulin chains (VH and VL), bound into a single polypeptide chain by a flexible 5-amino-acid linker (diabody format). Two molecules of human IL-2 are fused to the C terminus of each light chain. (B) The F16 antibody selectively targets the alternatively spliced A1 domain of tenascin-C (TnC-A1), which is spliced into the TnC molecule during angiogenesis and tissue remodeling. (C-D) Pharmacokinetic F16IL2 concentration/time curves in the serum of patients from cohorts 1 and 2 vs cohorts 3 and 4. Data are presented as mean ± standard error of the mean (SEM) on a linear (C) and a logarithmic (D) scale. Three patients were excluded: UPN004 (anomalous low values caused by technical problems), UPN006 (insufficient PK sampling), and UPN901 (stability issue with serum samples). (E) Acute body temperature changes after infusion of F16IL2 on day 1 and BI 836858 on day 3. Fevers were the most frequent treatment-related AEs in the study and body temperature elevations were predominantly observed within hours after the first administration of F16IL2.
The F16IL2 immunocytokine and its ECM target TnC-A1, F16IL2 pharmacokinetics, and body temperature changes after F16IL2 and BI 836858 administration. (A) The F16 antibody fragment is composed of variable regions of human heavy and light immunoglobulin chains (VH and VL), bound into a single polypeptide chain by a flexible 5-amino-acid linker (diabody format). Two molecules of human IL-2 are fused to the C terminus of each light chain. (B) The F16 antibody selectively targets the alternatively spliced A1 domain of tenascin-C (TnC-A1), which is spliced into the TnC molecule during angiogenesis and tissue remodeling. (C-D) Pharmacokinetic F16IL2 concentration/time curves in the serum of patients from cohorts 1 and 2 vs cohorts 3 and 4. Data are presented as mean ± standard error of the mean (SEM) on a linear (C) and a logarithmic (D) scale. Three patients were excluded: UPN004 (anomalous low values caused by technical problems), UPN006 (insufficient PK sampling), and UPN901 (stability issue with serum samples). (E) Acute body temperature changes after infusion of F16IL2 on day 1 and BI 836858 on day 3. Fevers were the most frequent treatment-related AEs in the study and body temperature elevations were predominantly observed within hours after the first administration of F16IL2.
In a recently completed phase 1 study, we have shown that up to 50 × 106 IU F16IL2 can be safely administered to AML patients relapsing after transplantation in combination with very low-dose cytarabine.21 F16IL2 treatment was associated with an expansion of immune effectors, particularly NK cells and showed early signs of clinical activity in heavily pretreated patients, but responses lasted only weeks to a few months. We hypothesized that an ADCC-evoking mAb would be an attractive combination partner for F16IL2 by enhancing leukemia recognition and redirecting the cytotoxic potential of allogeneic NK cells to leukemic blasts. Indeed, in preclinical experiments, IL-2-based immunocytokines potently boosted the activity of ADCC-inducing mAbs,18,22,23 but a clinical evaluation of this concept has not yet been pursued.
BI 836858 is a human anti-CD33 IgG1 mAb, which is Fc-engineered for increased binding to FcγRIIIA/CD16.24 It binds with low nanomolar affinity to CD33, which is expressed in most AML cases25 and displays decelerated internalization kinetics compared with previously developed CD33 mAbs. In vitro, BI 836858 has potent activity against AML blasts via ADCC mediated by autologous or allogeneic NK cells.24 A first-in-human dose-escalation trial with BI 836858 (registered on clinicaltrials.gov as NCT01690624) in 27 patients with relapsed/refractory AML (26% had a prior allogeneic HSCT) has been published recently.26 Although BI 836858 showed a manageable safety profile at doses of 10 to 40 mg/wk, and the maximum tolerated dose (MTD) was not reached, no objective responses were observed, possibly because of the low levels of baseline effector cells in this patient population.
We hypothesized that the F16IL2-mediated anchoring of IL-2 signals to the leukemia microenvironment would increase NK-cell abundance and activity to potentiate anti-CD33 ADCC responses at the site of disease and investigated this in a novel-novel combination phase 1 trial for AML relapse after HSCT.
Patients and methods
Patients
Patients aged 18 to 75 years with AML relapse after allogeneic HSCT were eligible. Relapse was defined as the reemergence of at least 5% BM blasts, the appearance of blasts in peripheral blood (PB) or histologically proven extramedullary relapse. Patients had to have an Eastern Cooperative Oncology Group performance status ≤2, adequate renal and hepatic function, no known central nervous system leukemia, no prior treatment with anti-CD33 mAbs, no active graft-versus-host disease (GVHD) requiring systemic immunosuppression (unless controlled with low-dose steroids) and no previous treatment within 4 weeks or a minimum of 5 half-lives of the treatment for the current AML relapse (except hydroxyurea).
Study design
This study was an open-label, single-arm, dose-escalation phase 1 trial conducted at 2 sites (Münster and Dresden) to find and investigate a safe dose of F16IL2 and BI 836858 in patients with posttransplant AML relapse. Dose escalation was guided by a Bayesian logistic regression model fitted to binary toxicity outcomes, with provisional dose levels (DLs) of 10 × 106 IU and 20 × 106 IU for F16IL2, and 10, 20, 40, 80, 160, and 320 mg for BI 836858 (supplemental Methods).
F16IL2 was given on days 1, 8, 15, and 22 of a 28-day cycle via rate-controlled IV infusion over 3 hours after premedication with paracetamol or metamizole. BI 836858 was given IV on days 3, 10, 17, and 24 over up to 5 hours. By protocol amendment, from cohort 4 onward, the first dose of BI 836858 was split, with a fixed dose of 20 mg administered on day 3 and the remaining dose administered on day 4. Premedication for BI 836858 included prednisolone (100 and 50 mg before the first and second administration, respectively, could be omitted from the third administration in the absence of infusion-related reactions), paracetamol, and an antihistamine equivalent to 50 mg diphenhydramine (further concomitant antimicrobial prophylaxis or treatment and unscheduled steroids are listed in supplemental Table 2). Response evaluation was performed after each cycle. Patients were treated until a maximum of 6 cycles, dose-limiting toxicity (DLT), AML progression or relapse, or treatment discontinuation requested by the patient or the investigator.
The protocol (EudraCT 2015-004763-37) was approved by the Paul Ehrlich Institute, the ethics committee of the Westphalian Wilhelms-University Münster, and the Chamber of Physicians Westphalia-Lippe, in accordance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines. Investigators obtained written informed consent from all patients before performing study-specific procedures. The manuscript was written solely by the authors without third-party assistance.
Study end points
The primary end point was to assess the safety profile, including MTD and recommended dose of F16IL2 combined with BI 836858. Safety assessments included surveillance of adverse events (AEs), vitals, physical examination, laboratory tests, electrocardiograms, and echocardiography. The DLT period was the first treatment cycle (28 days).
Secondary end points were efficacy, pharmacokinetics (PK), pharmacodynamics, and immunogenicity of F16IL2 combined with BI 836858. Efficacy was assessed after each cycle by modified revised International Working Group criteria.27 Extramedullary AML was followed by imaging procedures according to institutional guidelines.
PK
PK analyses were performed as previously described.28 In brief, serum was frozen and later batch-analyzed to measure levels of F16IL2 by enzyme-linked immunosorbent assay, with TnC-A1 used for plate coating and a mouse anti-IL-2 mAb used for detection of TnC-A1-bound F16IL2. A bicompartment model was used.
PK properties of BI 836858 were presented in a recent phase 1 trial26 and were not analyzed for this study.
Human anti–fusion protein antibodies
Immunogenicity of F16IL2 was tested with a validated enzyme-linked immunosorbent assay, as published,28 with modifications as detailed in supplemental Methods.
Immune effector cell analyses
Lymphocyte phenotypes in BM and PB were serially investigated by flow cytometry during therapy using antibodies against CD3, CD4, CD8, CD16, CD25, CD45RO, CD45RA, CD56, CD127, CD197, CD279, CD335 (NKp46), CD337 (NKp30), and γδTCR (BD Pharmingen), as previously described.21 Gating strategies are depicted in supplemental Figure 5. A complete blood count on the same day as flow analysis provided percentage of lymphocytes and an absolute lymphocyte count to calculate the absolute number of cells.
Statistical analysis
Study measures were summarized by descriptive statistics. AEs and laboratory abnormalities were based on Common Terminology Criteria for Adverse Events v.4.03 and were summarized by type, frequency, severity, relatedness, and seriousness. All patients who received any amount of F16IL2 or BI 836858 were included in the safety analyses. The efficacy-evaluable analysis set included all patients who received any amount of either antibody, had at least 1 postbaseline disease assessment (BM or imaging), or discontinued treatment because of clinically progressive disease (PD).
Overall survival (OS) was calculated by the Kaplan-Meier estimator. To evaluate the association of response with survival, OS was measured from a 28-day landmark (end of C1) and response was introduced as a time-dependent covariable. Time-dependent association was evaluated in a Cox proportional hazard model, plotted according to the Simon-Makuch method and compared with the Mantel-Byar test. Survival analyses were performed using the statistical environment R, version 4.1.1. Flow cytometry data were analyzed using a mixed effects analysis of variance model with Holm-Šidák corrections for multiple comparisons using GraphPad Prism (version 9.2.0).
Results
Patient characteristics
From December 2016 through March 2020, 19 patients were screened, and 15 patients with a median age of 50 (range, 20-68) years were enrolled. Baseline characteristics are summarized in Table 1. All patients had CD33+ AML (supplemental Figure 1). Four patients entered the study after 2 and 2 patients after 3 allogeneic transplants. Three patients with extramedullary manifestations were followed by 18F-fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG-PET/CT). Subjects were treated across 4 dosing cohorts: cohort 1 (10 × 106 IU F16IL2/10 mg BI 836858; n = 5), cohort 2 (10 × 106 IU/20 mg; n = 3), cohort 3 (20 × 106 IU/20 mg; n = 4), and cohort 4 (20 × 106 IU/40 mg; n = 3) (supplemental Figure 2). All subjects were enrolled within the dose escalation part and a planned expansion cohort was not pursued.
Safety
Of 15 patients, 9 received 1 cycle and 6 received 2 cycles of therapy. Reasons for study termination were refractory/progressive disease (n = 9), DLT (n = 2), AE/severe AE (SAE) (n = 2), HSCT after response to study treatment (n = 1), and patient’s decision (n = 1). No patient died within the first 30 days of treatment.
In total, 200 AEs of any grade were reported, and all patients experienced AEs, consistent with a cohort of patients with posttransplant AML relapse. AEs observed in >1 patient are summarized in Table 2 (complete list in supplemental Table 1). The most frequent drug-related AEs (F16IL2 or BI 836858 or combination) were pyrexia (87%), chills (80%), and infusion-related reactions (60%), consistent with the expected toxicity profile of cytokine-armed and naked mAbs. These events were all manageable, transient, and of grade ≤2. F16IL2 was typically associated with body temperature elevations within the first hours after infusion, which tended to persist until administration of BI 836858 2 days later, given after premedication with steroids (Figure 1E; supplemental Figure 3A). Whereas F16IL2-induced fevers were frequently observed upon first administration, they were less pronounced in response to subsequent infusions (supplemental Figure 3A). No relevant cytokine-induced hypotension was observed (supplemental Figure 3B-C). The most common non-infusion–related AEs were nausea (60%) and vomiting (33%). Drug-related AEs grade ≥3 were reported in 5 patients and included cytopenias, febrile neutropenia, acute GVHD, increased liver enzymes, dyspnea, hypoxia, cough, pulmonary edema, hypokalemia, and hypophosphatemia (Table 1).
Cumulatively, 15 SAEs were reported in 8 patients; 3 of them considered drug-related: cytokine-release syndrome with fever and chills (UPN909 in cohort 3, mismatched unrelated donor [MMUD], related to BI 836858), pulmonary edema (UPN007 in cohort 3, haploidentical related donor [HRD], related to F16IL2), and acute gastrointestinal and skin GVHD (UPN012 in cohort 4, matched unrelated donor [MUD], related to both drugs; supplemental Table 1). One DLT occurred at each of DL 3 (pulmonary edema in patient UPN007, HRD) and 4 (acute GVHD in patient UPN012, MUD). Two additional patients terminated treatment related to disease-related SAEs: infection grade 3 and cerebral hemorrhage grade 4 (after accidental fall).
Three patients (20%) developed acute GVHD: gastrointestinal GVHD in patients UPN008 (MMUD) and UPN009 (MUD) and gastrointestinal and skin GVHD in patient UPN012 (MUD). Patient UPN009 later developed chronic skin GVHD. One additional patient (UPN901, HRD) had a constant grade 2 liver GVHD before and after treatment.
No formal MTD was reached. The maximum tested dose was still considered safe. Thus, the MTD of F16IL2/BI 836858 is ≥20 × 106 IU/40 mg. Patient accrual was stopped at this DL because of a strategic company decision not to further develop BI 836858.
Pharmacokinetics
Cumulative F16IL2 serum concentration curves for cohorts 1 and 2 (10 × 106 IU) vs cohorts 3 and 4 (20 × 106 IU) are shown in Figure 1C-D. Maximum concentration (Cmax) level and area under the curve (AUC) revealed a dose-related exposure of patients to F16IL2 during the treatment period. Time to maximum concentration (Tmax) was shortly after the end of the 3-hour infusion of F16IL2 and cumulative Cmax was 35.7 ng/mL for 10 × 106 IU F16IL2 and 50.4 ng/mL for 20 × 106 IU F16IL2. AUC0-inf (ng*min/mL) was 30 068 for 10 × 106 IU F16IL2 and 87 899 for 20 × 106 IU F16IL2. Terminal half-life (t1/2) for 10 × 106 IU F16IL2 was 309 minutes and 537 minutes for 20 × 106 IU F16IL2 from the start of infusion. Analyses revealed some interindividual variation particularly in lower DLs: in some patients, preestimated Cmax dose levels were almost reached, and in others, lower-than-estimated Cmax values were measured. Similar observations were reported for other immunocytokines29 and could be interpreted as a consequence of trapping (for example, in the liver of some individuals, as observed with 131I-labeled small immunoproteins).30
PK properties of BI 836858 have been published recently,26 showing a dose-proportional increase of maximum plasma concentrations from 10 to 40 mg BI 836858 and a t1/2 of 11.3 (10 mg) to 34.5 hours (40 mg) in patients with relapsed/refractory AML.
Immunogenicity
Although F16IL2 is a fully human fusion protein, 34 samples collected from 15 patients at baseline, after C1 and end of treatment were screened for the presence of HAFA. No patient developed HAFA responses against F16IL2 (supplemental Figure 4).
Immune effector cell modulation
We serially investigated changes in lymphocyte subsets and activation markers in BM (n = 11) and PB (n = 12) before and during combination therapy. Overall, the most prominent alterations were observed in the NK-cell fraction (Figure 2A). Study therapy was associated with a significant increase in percentages of CD3−/CD56+ NK cells in BM and PB (Figure 2B-D). Similarly, also absolute NK-cell numbers steadily expanded during treatment, reaching mean BM levels of more than 700/µL and 1600/µL at the end of C1 and C2, respectively (Figure 2E).
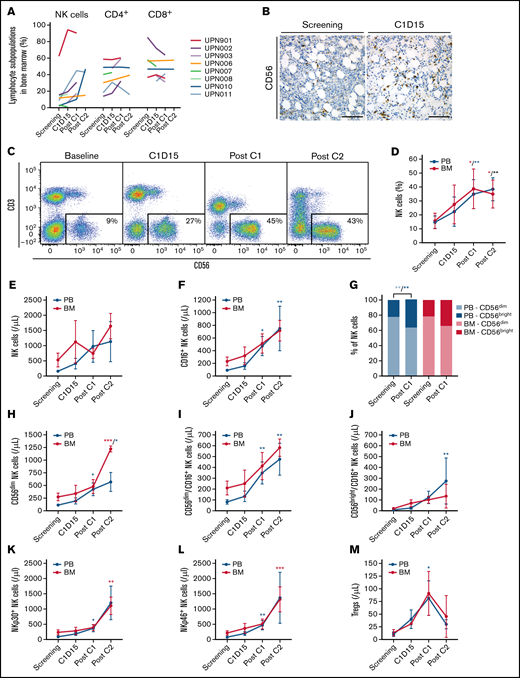
F16IL2 and BI 836858 combination therapy expands and activates NK cells. (A) Lymphocyte subsets in BM and PB were analyzed using flow cytometry at different time points during study treatment. Lymphocytes were immunophenotypically defined as CD45+/FSClow (for gating strategy see supplemental Figure 5). The most significant changes were observed in the NK-cell compartment. CD56 immunohistochemical stainings of pretherapeutic and posttherapeutic BM biopsies (UPN004) (B) and CD56 vs CD3 flow cytometry plots (UPN011) (C) illustrate progressive expansion of NK cells in BM during F16IL2 and BI 836858 combination therapy in 2 representative patients. (D) NK cell percentages (of lymphocytes) significantly increased over time in BM and PB. (E) Absolute number of NK cells in BM and PB during combination therapy. (F) F16IL2 and BI 836858 combination therapy resulted in a significant expansion of FcγRIIIA/CD16+ NK cells capable of mediating ADCC. (G) Ratios of CD56dim vs CD56bright NK cells indicated a shift toward CD56bright cells during combination therapy, but also CD56dim NK cells increased in absolute numbers (H). (I-J) CD56dim and CD56bright subsets expressing FcγRIIIA/CD16+ significantly expanded. (K-L) Number of NK cells expressing the natural cytotoxicity receptors NKp30 or NKp46 significantly increased over time. (M) Absolute numbers of Tregs peaked at the end of C1, with a subsequent decline during C2. P values indicate significant changes from values at screening as follows (mixed effects analysis of variance with Holm-Šidák corrections): *P < .05; **P < .01; ***P < .001. Data are presented as the mean ± standard error of the mean.
F16IL2 and BI 836858 combination therapy expands and activates NK cells. (A) Lymphocyte subsets in BM and PB were analyzed using flow cytometry at different time points during study treatment. Lymphocytes were immunophenotypically defined as CD45+/FSClow (for gating strategy see supplemental Figure 5). The most significant changes were observed in the NK-cell compartment. CD56 immunohistochemical stainings of pretherapeutic and posttherapeutic BM biopsies (UPN004) (B) and CD56 vs CD3 flow cytometry plots (UPN011) (C) illustrate progressive expansion of NK cells in BM during F16IL2 and BI 836858 combination therapy in 2 representative patients. (D) NK cell percentages (of lymphocytes) significantly increased over time in BM and PB. (E) Absolute number of NK cells in BM and PB during combination therapy. (F) F16IL2 and BI 836858 combination therapy resulted in a significant expansion of FcγRIIIA/CD16+ NK cells capable of mediating ADCC. (G) Ratios of CD56dim vs CD56bright NK cells indicated a shift toward CD56bright cells during combination therapy, but also CD56dim NK cells increased in absolute numbers (H). (I-J) CD56dim and CD56bright subsets expressing FcγRIIIA/CD16+ significantly expanded. (K-L) Number of NK cells expressing the natural cytotoxicity receptors NKp30 or NKp46 significantly increased over time. (M) Absolute numbers of Tregs peaked at the end of C1, with a subsequent decline during C2. P values indicate significant changes from values at screening as follows (mixed effects analysis of variance with Holm-Šidák corrections): *P < .05; **P < .01; ***P < .001. Data are presented as the mean ± standard error of the mean.
NK cells expressing FcγRIIIA/CD16 are those capable of mediating ADCC. CD16+ NK cells cumulatively increased with ongoing treatment (Figure 2F). NK cells can be further divided into CD56dim and CD56bright subsets. CD56dim NK cells are considered naturally more cytotoxic than CD56bright cells. In turn, CD56bright NK cells have the capacity for high-level cytokine production and a lower cytotoxicity at rest, but may exert similar levels of cytotoxicity after cytokine activation.31,32 Whereas the ratio of CD56dim and CD56bright NK cells slightly shifted toward CD56bright cells during treatment (Figure 2G), also CD56dim cells increased in absolute number in BM and PB (Figure 2H). Both CD56dim/CD16+ and CD56bright/CD16+ NK cells significantly expanded over time, with increases ranging between 2-fold (CD56dim/CD16+ cells in BM) and 18-fold in the initially small CD56bright/CD16+ subset in PB at the end of C1 compared with pretreatment levels (Figure 2I-J). Finally, treatment was associated with a steady expansion of NK cells expressing the activating natural cytotoxicity receptors NKp30 or NKp46 (Figure 2K-L).
Percentages of CD4+ or CD8+ T cells and their effector phenotypes were not significantly altered (Figure 2A; supplemental Figure 6). However, the number of T cells with Treg phenotype (CD4+/CD25high/CD127−) increased after therapy (Figure 2M), peaking after C1, with a subsequent decline during C2, as previously observed with continued low-dose IL-2 treatment.33-36
Clinical response
Three of 15 patients (20%) had an objective response (Figure 3A): 1 CR, 1 CR with incomplete count recovery (CRi), and 1 partial remission (PR) in extramedullary AML. Another 4 patients had stable disease (SD). No formal responses occurred at the 2 starting DLs. In patients treated at the 2 highest DLs, the response rate (CR/CRi/PR) was 43%. Baseline and outcome characteristics of responding patients are described in the following case vignettes and in supplemental Table 3.
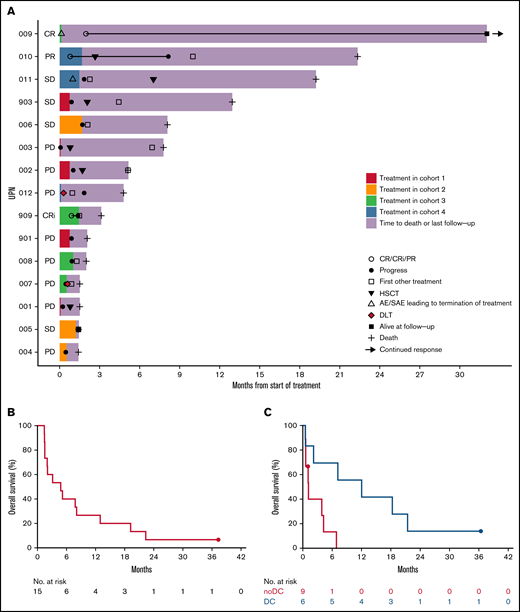
Antileukemic efficacy of F16IL2 and BI 836858 combination therapy. (A) Swimmer plot of individual treatment courses and outcome in all 15 patients. Cohort assignment is color coded and AEs, SAEs, or DLTs leading to treatment termination are indicated. Antileukemic activity was mainly observed in the 2 highest DLs. (B) Kaplan-Meier estimates of OS of all 15 patients treated in the study. Median OS was 4.8 (interquartile range, 1.5-12.9) months, with a 6- and 12-month OS of 40% and 27%, respectively. (C) Simon-Makuch estimates of OS by response with a 28-day landmark (end of C1), using response as a time-dependent covariable. Among those 7 patients in whom AML was at least temporarily controlled with study treatment, median OS was 12.0 months (vs 1.2 months in nonresponders), with a 12-month OS of 56% vs 0% (hazard ratio, 0.17; 95% confidence interval, 0.03-0.84; Mantel-Byar test, P = .016). DC, disease control (SD or better).
Antileukemic efficacy of F16IL2 and BI 836858 combination therapy. (A) Swimmer plot of individual treatment courses and outcome in all 15 patients. Cohort assignment is color coded and AEs, SAEs, or DLTs leading to treatment termination are indicated. Antileukemic activity was mainly observed in the 2 highest DLs. (B) Kaplan-Meier estimates of OS of all 15 patients treated in the study. Median OS was 4.8 (interquartile range, 1.5-12.9) months, with a 6- and 12-month OS of 40% and 27%, respectively. (C) Simon-Makuch estimates of OS by response with a 28-day landmark (end of C1), using response as a time-dependent covariable. Among those 7 patients in whom AML was at least temporarily controlled with study treatment, median OS was 12.0 months (vs 1.2 months in nonresponders), with a 12-month OS of 56% vs 0% (hazard ratio, 0.17; 95% confidence interval, 0.03-0.84; Mantel-Byar test, P = .016). DC, disease control (SD or better).
In cohort 3, a 68-year-old woman (UPN009) with primary refractory AML with complex karyotype (del(5),del(7),−17,del(18)) was enrolled 4 months after HSCT. BM evaluation, performed because of a decline in PB donor cell chimerism, revealed a relapse with the leukemia-associated immunophenotype (SSClow/mid/CD45dim/CD13−/CD33+/CD56+/CD117+). Combination treatment with F16IL2 and BI 836858 was prematurely stopped because of a subdural hematoma after an accidental fall that necessitated neurosurgical intervention. After surgery, the patient’s condition gradually improved, and she subsequently achieved CR without further leukemia-specific therapy. At this writing, CR with full donor chimerism was ongoing for 35 months.
At screening, patient UPN909 had 91% BM blasts showing the preknown leukemia-associated immunophenotype (CD13−/CD33+/HLA-DR−/CD34+/CD56+/CD117−) and cytogenetic aberrations (t(?;11q23.3), del(9)). He was treated in cohort 3 and achieved a CRi after C1. Fifteen days later, the patient experienced PD because of the occurrence of central nervous system leukemia. He received intrathecal chemotherapy but died 2 months later.
In cohort 4, 27-year-old male patient UPN010 entered the study with widespread extramedullary AML manifestations after 2 transplants (Figure 4). There was no overt BM relapse, but donor cell chimerism in sorted CD34+ BM cells had decreased to 89% before treatment initiation. After C1, he achieved a PR with a near complete disappearance of extramedullary lesions and regained complete BM CD34+ chimerism (99%). Interestingly, infusion of F16IL2 was associated with pain in different chloroma lesions, indicative of effective targeting. Study treatment enabled a third HSCT, but AML relapse was diagnosed 8 months after enrollment in the study.
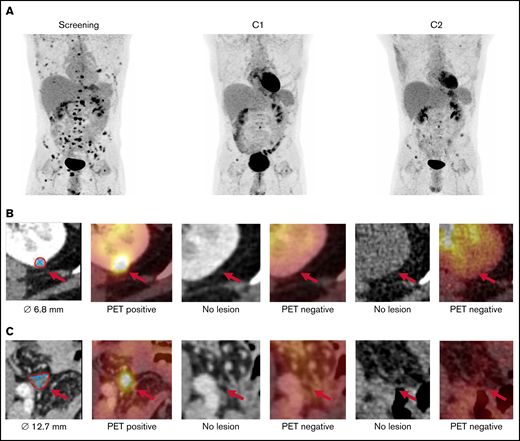
Whole-body 18F-FDG-PET/CT in patient UPN010 with disseminated extramedullary AML after 2 allogeneic transplants. Maximum intensity projection (A) and transaxial CT and fused PET/CT images of 2 exemplary lesions at screening, after C1 and after C2: left kidney (B) and lower pelvic cavity (C).
Whole-body 18F-FDG-PET/CT in patient UPN010 with disseminated extramedullary AML after 2 allogeneic transplants. Maximum intensity projection (A) and transaxial CT and fused PET/CT images of 2 exemplary lesions at screening, after C1 and after C2: left kidney (B) and lower pelvic cavity (C).
Median OS among all 15 patients was 4.8 (interquartile range, 1.5-12.9) months, with a 6- and 12-month OS of 40% and 27%, respectively (Figure 3B). Among those 7 patients whose AML was at least temporarily controlled with study treatment (SD or better), the hazard risk of death was less than 1:5 compared with that of nonresponders, when introducing response in a Cox proportional hazards regression as a time-dependent covariable (hazard ratio, 0.17; 95% CI, 0.03-0.84; Mantel-Byar test, P = .016). Median OS was 12.0 vs 1.2 months, with a 12-month OS of 56% vs 0% (Figure 3C). Patients achieving SD or better tended to have a longer time from HSCT to study treatment than nonresponders (median, 14.0 vs 4.5 months; P = .4). All but the 1 patient in CR had died.
GVHD developed in 1 of 3 responding patients (UPN009 with CR, MUD) and in 2 of 12 patients without a formal response (UPN008, MMUD; UPN012, MUD).
Discussion
The F16IL2 fusion was designed to selectively localize IL-2 to TnC-A1, an acellular yet highly tumor-specific antigen.15 -17,20,37 In our early-phase study, we hypothesized that F16IL2 would convert the BM into an immunologically “hotter” microenvironment to amplify anti-CD33-mAb–mediated ADCC. CD33 is an attractive target for ADCC-mediating mAbs because it is expressed on leukemic blasts in most AML patients,25 as in this study.
Posttransplant AML is a delicate indication for the development of engineered cytokine products. On the one hand, the allogeneic immune context carries a potential risk of exacerbating toxicity from cytokine effects. Thus, the chosen starting doses of both immune-based study drugs were relatively low, even though F16IL2 had previously been applied at up to 50 × 106 IU with very low-dose cytarabine.21 In a trial of nivolumab for relapsed hematologic malignancies after allogeneic HSCT, deescalation to a dose as low as 0.5 mg/kg (compared with fixed doses of 240 or 480 mg, used outside the transplantation context) was necessary, and still substantial toxicities occurred from GVHD and other immune-related AEs.38 On the other hand, immunotherapies classically require a longer time to elicit antileukemic effects, which is barely compatible with the clinical condition of patients relapsing after transplantation.39
The most common treatment-emerging AEs were related to the infusion of either antibody or to direct biological effects of the IL-2 cargo in circulation (infusion-related reactions, chills, and pyrexia). Pain in chloroma lesions and recurrent fevers for days after administration provide indirect evidence supporting both the targeting properties of the immunocytokine and its on-target persistence. It was noticeable that fevers were most pronounced after the first administration of F16IL2 and typically persisted until administration of BI 836858 on day 3. Premedication of BI 836858 included a single dose of 100 mg prednisolone, which may have mitigated the inflammatory effects of F16IL2 given 2 days before. Indeed, when F16IL2 was used in combination with very-low-dose cytarabine, drug-induced fevers tended to persist longer.21 In line with this observation, significant changes in T-cell frequencies and their subsets were only observed when F16IL2 was combined with very-low-dose cytarabine (without the need for steroid premedication) in the above-mentioned previous trial21 and were not detected in this study. Whereas pulmonary edema occurred as the DLT related to F16IL2 in a patient with a haploidentical graft, overall, there was no obvious correlation between donor type and toxicity.
F16IL2 treatment led to a marked expansion of NK cells. These observations reproduced preclinical findings, which revealed a dominant role of NK cells for the therapeutic activity of IL2-based immunocytokines.20 Although the ratio of CD56dim to CD56bright subpopulations shifted toward CD56bright NK cells, cytotoxic CD56dim and ADCC-competent CD56dim/CD16+ NK cells significantly increased in absolute numbers with continued therapy. Interestingly, also the initially small CD56bright/CD16+ subset, which has been described as highly functional and competent to elicit ADCC,40 expanded over time. However, given the multiple mechanisms of NK effector functions, the relative contribution of ADCC, the cytotoxicity via the missing-self mechanism, and the modulation of T-cell activity through cytokine production to the overall therapeutic effect is difficult to dissect.6 As analyses of immune effector cell modulation are based on a small data set, they may be skewed in favor of patients achieving a response or at least disease control and therefore receiving more than 1 cycle of treatment. Furthermore, evaluable pretherapeutic and postherapeutic sample pairs were available in only 1 of 3 patients with a formal response. Because of these limitations, pharmacodynamic data have to be interpreted with caution, and we did not attempt to correlate NK-cell modulation with clinical efficacy.
We found that tolerable doses of F16IL2 and BI 836858 induced clinically significant responses in this heavily pretreated cohort of patients with AML, including 1 durable CR. Although efficacy was not the main end point and no objective responses were seen at the 2 lower DLs, 3 of 7 patients (43%) who received 20 × 106 IU F16IL2 along with 20 or 40 mg BI 836858 had a response, including patients with extramedullary disease or high-risk cytogenetics. Among those who achieved at least disease stabilization, a significant OS advantage was observed. However, rapidly proliferating peripheral blasts in patients treated at the lower DLs were not efficiently controlled. Concomitant hydroxyurea was not allowed by protocol but should be part of future studies involving F16IL2 to facilitate delayed therapeutic effects, as seen with other immunotherapies. When combining evidence from this and previous trials,21,41 posttransplant extramedullary AML, which is frequently refractory to standard therapies, seems to be particularly responsive to F16IL2. Four of 5 evaluable patients achieved some degree of chloroma shrinkage in response to F16IL2-based immunotherapy. A particular sensitivity of posttransplant extramedullary AML has also been described with anti-CTLA-4 therapy.42
A more theoretical limitation of our study is the use of a cytokine payload with potential double-edged activity. Apart from stimulating effector cells, IL-2 controls the growth of Tregs, especially at low doses that preferentially activate trimeric IL-2 receptors.8 Ultra–low-dose IL-2 has been suggested for treatment of autoimmune and alloimmune disorders, including GVHD,33,34,43,44 even though others have questioned the concept of IL-2–mediated control of inflammation in the particular setting of allogeneic transplantation.45 In our study, not only NK cells but also Tregs increased in response to study treatment. Similarly, in a recent trial, decitabine combined with G-CSF led to a concurrent expansion of NK, CD8+ T, and Treg cells and reduced the incidence of AML relapse after transplantation without promoting GVHD.46 Interestingly, it has been shown that the BM stroma may mediate a conditional neutralization of Tregs in the BM via IL-1beta and IL-6, providing a favorable environment for antitumor responses.47 In other preclinical models, Tregs have been shown to suppress GVHD without decreasing GVL effects.48 Nevertheless, it is entirely possible that concomitant Treg expansion attenuated further effector cell activation in our study and prevented development of more severe GVHD at the same time. Overall, it is not obvious that alternative cytokines with reduced Treg binding would constitute better payloads for BM-directed mAbs after transplantation. In fact, in preclinical xenogeneic transplantation, development of lethal GVHD was associated with administration of IL-15 but not IL-2.49 Unfortunately, we were unable to measure the suppressive activity of phenotypic Tregs.
In summary, in the difficult-to-treat situation of posttransplant AML relapse, responses to F16IL2 and BI 836858 combination therapy were observed at higher DLs. Although clinical development of BI 836858 is not further pursued, this trial may serve as a starting point for a broader evaluation of ECM-anchoring immunocytokines as versatile mutation- and surface marker–agnostic tools to boost on-target activity of mAbs or other immunological maneuvers (eg, NK cell engagers) in AML.
Acknowledgments
The authors thank the patients, their families, and their caregivers for participating in this trial and the study teams at Münster and Dresden.
This work was supported by Philogen S.p.A., Siena, Italy, and Boehringer Ingelheim Pharma, Biberach, Germany.
Authorship
Contribution: W.E.B., B.R., T.H., M.M., D.N., and C.S. conceived and designed the study; C. Röllig, L.R., C.S., J.-H.M., M.W., K.W., T.K., M. Stelljes, M.B., W.E.B. recruited and managed patients; B.A., C. Rossig, A.F.B., and C.S. performed and analyzed flow cytometry analyses; M.G., D.H., and M. Schäfers performed imaging studies; W.H. performed immunohistochemistry; A.F.B., L.A., A.S., G.L., J.-H.M., M. Stelljes, T.H., D.N., W.E.B., and C.S. analyzed the data; A.F.B. and C.S. wrote the manuscript; and all authors participated in data interpretation, reviewed and revised the manuscript, and approved the final manuscript.
Conflict-of-interest disclosure: D.N. is a board member of and has ownership interest in Philogen. T.H. and M.M. are employees of Philogen. W.E.B. is a member of the scientific advisory board of Philogen, is a shareholder of Philogen, and receives honoraria and travel grants from Philogen. M. Schäfers received an institutional grant on preclinical testing of a novel ligand outside the scope of this study from Philogen. The remaining authors declare no competing financial interests.
Correspondence: Christoph Schliemann, Department of Medicine A, University Hospital Münster, Albert-Schweitzer-Campus 1, 48149 Münster, Germany; e-mail: christoph.schliemann@ukmuenster.de.