

Key Points
Patients with OM-CMML presented longer OS and acute myeloid leukemia–free survival than did those with D-CMML or P-CMML.
The progression of a high proportion of OM-CMML into D-CMML and D-CMML into P-CMML reinforces the evolutionary continuum of CMML.

Abstract
Patients with oligomonocytic chronic myelomonocytic leukemia (OM-CMML) are currently classified according to the 2017 World Health Organization myelodysplastic syndromes classification. However, recent data support considering OM-CMML as a specific subtype of chronic myelomonocytic leukemia (CMML), given their similar clinical, genomic, and immunophenotypic profiles. The main purpose of our study was to provide survival outcome data of a well-annotated series of 42 patients with OM-CMML and to compare them to 162 patients with CMML, 120 with dysplastic type (D-CMML), and 42 with proliferative type (P-CMML). OM-CMML had significantly longer overall survival (OS) and acute myeloid leukemia–free survival than did patients with CMML, considered as a whole group, and when compared with D-CMML and P-CMML. Moreover, gene mutations associated with increased proliferation (ie, ASXL1 and RAS-pathway mutations) were identified as independent adverse prognostic factors for OS in our series. We found that at a median follow-up of 53.47 months, 29.3% of our patients with OM-CMML progressed to D-CMML, and at a median follow-up of 46.03 months, 28.6% of our D-CMML group progressed to P-CMML. These data support the existence of an evolutionary continuum of OM-CMML, D-CMML, and P-CMML. In this context, we observed that harboring more than 3 mutated genes, carrying ASXL1 mutations, and a peripheral blood monocyte percentage >20% significantly predicted a shorter time of progression of OM-CMML into overt CMML. These variables were also detected as independent adverse prognostic factors for OS in OM-CMML. These data support the consideration of OM-CMML as the first evolutionary stage within the proliferative continuum of CMML.
Introduction
Oligomonocytic chronic myelomonocytic leukemia (OM-CMML) is defined as those myelodysplastic syndromes (MDSs) or unclassifiable myelodysplastic/myeloproliferative neoplasms with relative monocytosis (≥10% monocytes) and a monocyte count of 0.5 × 109/L to <1 × 109/L.1,2 Recent data support the consideration of OM-CMML as a specific subtype of chronic myelomonocytic leukemia (CMML), because the clinical, morphological, cytogenetic, molecular, and immunophenotypic profiles are similar to those of patients with overt CMML.1,3,4 Given the overall good prognosis described in OM-CMML, it could be inferred that this group of patients represents a precondition in the early spectrum of dysplastic CMML (D-CMML).1,3,4 Although the disease progresses to overt CMML in a high percentage of patients, globally reinforcing the precondition concept, some patients die of evolution to acute myeloid leukemia (AML) or of bone marrow (BM) failure before progression to overt CMML.1,3,4 This fact is different from that observed in other well-accepted preconditions (eg, monoclonal gammopathy of undetermined significance and clonal hematopoiesis of indeterminate potential). Otherwise, it is important to assess whether the specific CMML prognostic factors5-12 applied to OM-CMML provide better predictive accuracy of outcome than classic MDS prognostic variables,13,14 given that most patients are currently categorized as having MDS according to the World health Organization (WHO) 2017 classification.3,15,16 Although most of the prognostic factors described in MDS are also consistent prognostic determinants in CMML, there are some that are specific to the latter (eg, trisomy 8 as a high-risk cytogenetic abnormality, and blast cell count defined as the sum of blasts and promonocytes).5-12 Of note, leukocytosis is a well-recognized independent prognostic factor in CMML,6,7,10,12 but it is not applicable to OM-CMML prognostic stratification, because the presence of ≥10 × 109/L leukocytes implies having at least 1 × 109/L monocytes and therefore meeting CMML diagnostic criteria. As previously reported, mutations in genes of the RAS pathway (eg, CBL, NRAS, and KRAS) are almost absent in OM-CMML, because they have been associated with proliferative features.1,3,4 Moreover, if present, these genes are more likely to constitute small clones in patients with OM-CMML.4 In this sense, CBL is the only gene mutated in a significantly lower proportion of OM-CMML vs CMML cases.1,3 Likewise, although there are no differences in the proportion of patients with OM-CMML carrying an ASXL1 mutation, when compared with those with D-CMML, this mutation is significantly overrepresented in proliferative CMML (P-CMML).3 Given the poor prognosis associated with RAS-pathway mutations,12 ASXL1 mutations,7,9 and leukocytosis,6,7,10,12 OM-CMML is expected to present a better outcome than the next steps in the proliferative continuum of CMML (ie, D-CMML and P-CMML). Regardless, OM-CMML may present other well-known adverse prognostic variables, such as an excess of blasts, because its presence does not invalidate the diagnosis. In light of all of these facts, the purpose of this study was to provide survival data from a well-annotated series of 42 patients with OM-CMML, to compare with the survival data from 162 patients with CMML: 120 with D-CMML and 42 with P-CMML.
Methods
Patients
We retrospectively studied 204 patients, 42 of them had MDS meeting OM-CMML diagnostic criteria and 162 were diagnosed with CMML. All diagnostics were established according to WHO 2017 criteria.15,16
Of the 162 patients meeting CMML criteria, 120 were diagnosed with D-CMML and 42 with P-CMML: 34 with CMML-0, 70 with CMML-1, and 58 with CMML-2.
According to the WHO 2017 MDS classification,15,16 the 42 patients with OM-CMML were classified into the following categories: 1 MDS with single-lineage dysplasia, 18 MDS with multilineage dysplasia, 4 MDS with ring sideroblasts and single-lineage dysplasia, 9 MDS with ring sideroblasts and multilineage dysplasia, 9 MDS with excess blasts-1, and 1 MDS with excess blasts-2.
The main patient characteristics are summarized in Table 1. The study was approved by the Parc de Salut Mar Clinical Research Ethics Committee (2016/6768/I) and conducted according to the Declaration of Helsinki.
Morphological studies
At least, 2 BM and 1 peripheral blood (PB) May-Grünwald-Giemsa–stained smears were used for conducting the morphologic analysis. A Prussian blue–stained BM smear was used for assessing the percentage of ring sideroblasts, and a nonspecific esterase, usually α-naphthyl butyrate esterase, was used to better identify the monocyte lineage. In addition, we performed a BM biopsy with a detailed histologic examination in all those cases in which it was necessary to exclude certain differential diagnoses with clinical and biological characteristics close to CMML or OM-CMML (eg, essential thrombocythemia with monocytosis, prefibrotic primary myelofibrosis with monocytosis, primary myelofibrosis with monocytosis, and systemic mastocytosis with an associated CMML). The WHO 2017 proposals for establishing the diagnosis of MDS and CMML were followed strictly.15 A comprehensive explanation of the morphological assessment is given in the supplemental Material.
Next-generation sequencing
Next-generation sequencing data were available in 94 patients (Figure 1). We analyzed the full exonic regions of 25 genes associated with myeloid malignancies (ASXL1, CALR, CBL, CSF3R, DNMT3A, ETV6, EZH2, IDH1, IDH2, JAK2, KIT, KRAS, MPL, NRAS, PRPF8, RUNX1, SETBP1, SF3B1, SH2B3, SRSF2, STAG2, TET2, TP53, U2AF1, and ZRSR2). The NGS methodology has been previously described by our group3 and is summarized in the supplemental Material.
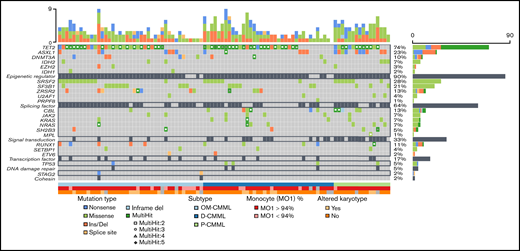
Next-generation sequencing mutational profile of patients with OM-CMML, D-CMML, and P-CMML.
Next-generation sequencing mutational profile of patients with OM-CMML, D-CMML, and P-CMML.
Flow cytometry analysis of monocyte subsets in PB
Multiparametric flow cytometry (FC) analysis of monocyte subsets was performed on whole PB collected into EDTA-coated tubes. Sample preparation, strategy of analysis, and the 5-tube experimental panel have been previously described by our group3 and are summarized in the supplemental Material. FC analysis of PB monocyte subsets has emerged as a useful diagnostic tool in CMML.3,17 -23 An increase in the classic monocyte (MO1) fraction to >94% shows high sensitivity and specificity for predicting CMML diagnosis.3,17-19,22 FC results are depicted in Table 1.
Statistical analysis
Categorical variables are described by frequencies and percentages and continuous variables as medians and ranges. For categorical data, comparisons of proportions were evaluated by χ2 test or Fisher’s exact test, as appropriate. For continuous variables, comparisons were assessed by nonparametric Mann-Whitney U or Kruskal-Wallis test, as appropriate. Survival curves were constructed by the Kaplan-Meier method, using the interval from the date of diagnosis to the date of last contact or death (overall survival [OS]), or AML evolution or death, whichever came first (AML-free survival [LFS]). Survival curves were compared by the log-rank test. We decided not to censor patients at the date of starting treatment, because only a small number of patients received disease-modifying treatments. Thirteen patients received hypomethylating agents (3 with OM-CMML, 8 with D-CMML, and 2 with P-CMML; χ2 = 0.886) and 3 patients received intensive chemotherapy followed by allogeneic stem cell transplantation (2 OM-CMML and 1 D-CMML; χ2 = 0.129). Univariate and multivariate analyses were performed using Cox’s proportional hazards model. The concordance index (C-index) for right-censored data was applied to assess the accuracy of the implemented models. To detect the best cutoff of time-to-CMML–prediction variables, we used the maximally selected rank statistics (Maxstat) method. We assessed the Spearman rank correlation to evaluate the strength of association between 2 variables. Differences were considered statistically significant when P < .05 in a 2-tailed test.
Results
Comparison of main characteristics of patients with OM-CMML, D-CMML, and P-CMML
The clinical findings in 42 patients with OM-CMML, 120 patients with D-CMML, and 42 patients with P-CMML are compared in Table 1. As shown, we observed no significant differences in age, sex, hemoglobin levels, platelet count, BM dysgranulopoiesis, BM dysthrombopoiesis, percentage of abnormal karyotypes, distribution of the Spanish cytogenetic risk groups,24 proportion of patients with mutations, and proportion of patients with a percentage of classic monocytes (MO1) >94%. Patients with OM-CMML had lower absolute leukocyte and monocyte counts, a predictable finding, given the definition of OM-CMML. However, they also had a lower percentage of PB and BM monocytes than did those with D-CMML and P-CMML. Although we did not observe differences when comparing OM-CMML with D-CMML, P-CMML had an inferior median percentage of dyserythropoiesis than OM-CMML. BM blast counts were higher in D-CMML and P-CMML when compared with OM-CMML. Likewise, the OM-CMML group had a lower proportion of patients categorized in higher-risk categories of the CMML 2017 WHO classification. P-CMML also presented a higher percentage of patients with PB blasts and a higher proportion of patients classified in higher-risk categories of the assessed prognostic scores. Finally, patients with P-CMML had a higher median number of mutated genes and a higher median number of mutations than those with OM-CMML or D-CMML.
The mutational profiles of OM-CMML, D-CMML, and P-CMML are depicted in Figure 1 and compared in Table 1. As shown, the only gene mutated in a significantly lower proportion of cases when OM-CMML was compared with D-CMML was CBL (2.4% vs 16.2%; P = .048). The P-CMML cases had a significantly higher proportion of CBL mutations than OM-CMML (31.3% vs 2.4%; P = .005) and a higher proportion of ASXL1 mutations than OM-CMML (62.5% vs 17.1%; P = .001) and D-CMML (62.5% vs 13.5%; P < .001). Moreover, the P-CMML group presented a significantly higher proportion of RAS-pathway mutations (ie, mutations in CBL, NRAS, and/or KRAS genes) than the OM-CMML (62.5% vs 4.9%; P < .001) and D-CMML (62.5% vs 27%; P = .014) groups. Likewise, D-CMML showed a significantly higher proportion of RAS-pathway mutations than OM-CMML (P = .010). Previous studies have shown that the allelic variant frequency (VAF) of CBL mutations is higher in P-CMML than in D-CMML.25 In our series, CBL mutation was observed in a single patient with OM-CMML (VAF, 2.8%); therefore, we could not perform this comparison among the 3 groups. However, patients with P-CMML presented a significantly higher VAF than those with D-CMML (median; interquartile range [IQR]), 40.5% [37.61-66.75] vs 2.9% [2.02-29.44]; P = .047). Our results are in line with those shown by Carr et al,25 who observed that NRAS mutations with VAF ≥30% and/or any other gene mutation of the RAS-pathway with VAF ≥19% were better predictors of the P-CMML phenotype than the presence of RAS-pathway mutations as a binary event.
OM-CMML has better outcomes than overt CMML
At a median follow-up of 48.07 months, 66.2% of patients had died (OM-CMML, median follow-up, 53.47 months, 38.1% deceased; D-CMML, median follow-up, 46.03 months, 69.2% deceased; P-CMML, median follow-up, 29.57 months, 85.7% deceased) and 18% of patients progressed to AML (9.5%, 16.7%, and 28.6% of patients with OM-CMML, D-CMML, and P-CMML).
Patients with OM-CMML had a significantly longer OS (median OS, 72.02 vs 39.43 months; P = .001) and LFS (median LFS: 72.02 vs 35.75 months; P = .001) than did those with CMML. In addition, patients with OM-CMML had a significantly longer OS than did those with D-CMML (median OS, 72.02 vs 50.37 months; P = .007) and P-CMML (median OS, 24.12; P < .001). Likewise, patients with OM-CMML had a significantly longer LFS than did those with D-CMML (median LFS, 72.02 vs 50.37 months; P = .008) and P-CMML (median LFS: 23; P < .001; Figure 2).
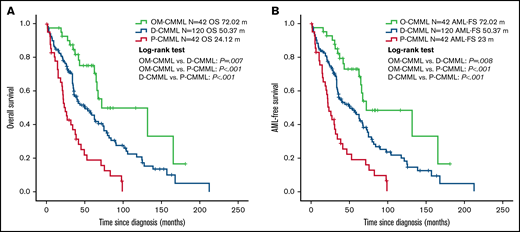
Overall survival and AML-free survival by Kaplan-Meier analysis of patients with OM-CMML, D-CMML, and P-CMML. (A) OS analysis. Patients with OM-CMML had a significantly longer OS than did those with D-CMML (median OS, 72.02 vs 50.37 months; P = .007) and P-CMML (median OS, 72.02 vs 24.12; P < .001). (B) LFS by Kaplan-Meier analysis of patients with OM-CMML, D-CMML, and P-CMML. Patients with OM-CMML had a significantly longer LFS than did those with D-CMML (median LFS, 72.02 vs 50.37 months; P = .008) and P-CMML (median LFS, 72.02 vs 23; P < .001). OS and LFS were compared with 2-sided log-rank tests.
Overall survival and AML-free survival by Kaplan-Meier analysis of patients with OM-CMML, D-CMML, and P-CMML. (A) OS analysis. Patients with OM-CMML had a significantly longer OS than did those with D-CMML (median OS, 72.02 vs 50.37 months; P = .007) and P-CMML (median OS, 72.02 vs 24.12; P < .001). (B) LFS by Kaplan-Meier analysis of patients with OM-CMML, D-CMML, and P-CMML. Patients with OM-CMML had a significantly longer LFS than did those with D-CMML (median LFS, 72.02 vs 50.37 months; P = .008) and P-CMML (median LFS, 72.02 vs 23; P < .001). OS and LFS were compared with 2-sided log-rank tests.
Univariate analyses for both OS and LFS, including OM-CMML and CMML patients, confirmed the utility of the assessed prognostic scores: CMML-specific prognostic scoring system (CPSS) (low/intermediate-1 vs intermediate-2/high categories; median OS: 63.28 vs 20.11; P < .001; median LFS: 63.28 vs 20.1; P < .001), CPSS-P (low/intermdiate-1 vs intermediate-2/high categories, median OS: 72.02 vs 22.77, P < .001; median LFS: 67.78 vs 21.36; P < .001), and Mayo prognostic model (low/intermediate vs high categories; median OS, 65.84 vs 19.02; P < .001; median LFS: 64.62 vs 18.4; P < .001).
Afterward, we applied the molecular CPSS (CPSS-mol) to 94 patients in our series for whom we had molecular data. CPSS-mol showed a good capacity to differentiate lower- from higher-risk patients in terms of OS (hazard ratio [HR], 3.5; 95% confidence interval [CI], 1.77-6.9; P < .001) and LFS (HR, 3.3; 95% CI, 1.7-6.42; P < .001).
Age was also detected as an adverse prognostic factor for OS (HR, 1.05; 95% CI, 1.03-1.076; P < .001) and LFS (HR, 1.045; 95% CI, 1.02-1.07; P < .001).
The better OS and LFS of patients with OM-CMML were retained after a subsequent multivariate adjustment by CPSS, CPSS-P, and the Mayo prognostic model (Table 2). The better outcomes of patients with OM-CMML were also retained when adjusted by each of these prognostic models and age (Table 3). Given that OM-CMML was detected as an independent protective prognostic factor, we assessed the impact of its addition to CPSS, CPSS-P, and the Mayo prognostic model. The addition of OM-CMML improved the accuracy for predicting OS of these 3 scores (C-index, 0.62, 0.64, and 0.67 for CPSS, CPSS-P, and the Mayo prognostic model, respectively; C-index: 0.66, 0.67, and 0.7 for CPSS+OM-CMML, CPSS-P+OM-CMML, and the Mayo prognostic model+OM-CMML, respectively). Later, we analyzed exclusively the OS of those patients classified into lower-risk categories of CPSS (ie, low and intermediate-1), CPSS-P (ie, low and intermediate-1), and the Mayo prognostic model (ie, low and intermediate). By considering the 3 proliferative stages (ie, OM-CMML, D-CMML, and P-CMML), lower-risk patients by CPSS and the Mayo prognostic model were split into 3 groups with significantly different OS. When selecting lower-risk patients by CPSS-P, patients with OM-CMML had longer OS than those with P-CMML, but we found no significant differences when compared with those with D-CMML (Figure 3).
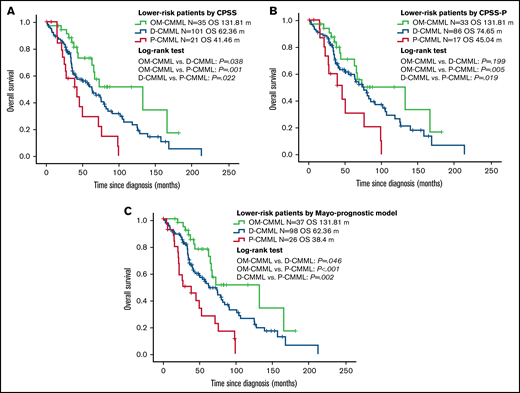
Overall survival by Kaplan-Meier analysis of lower-risk patients according to CPSS, CPSS-P, and Mayo-prognostic model. (A) OS analysis. Patients with OM-CMML had a significantly longer OS than did those with D-CMML (median OS, 131.81 vs 62.36 months; P = .038) and P-CMML (median OS, 131.81 vs 41.46; P = .001). (B) OS by Kaplan-Meier analysis of lower-risk patients by CPSS-P. Patients with OM-CMML did not have a significantly longer OS than did those with D-CMML (median OS: 131.81 vs 74.65 months; P = .199), but presented a significantly longer OS than did those with P-CMML (median OS, 131.81 vs 45.04; P = .005). (C) OS by Kaplan-Meier analysis of lower-risk patients by the Mayo prognostic model. Patients with OM-CMML had a significantly longer OS than did those with D-CMML (median OS, 131.81 vs 62.36 months; P = .046) and P-CMML (median OS, 131.81 vs 38.4; P < .001). OS was compared with 2-sided log-rank tests.
Overall survival by Kaplan-Meier analysis of lower-risk patients according to CPSS, CPSS-P, and Mayo-prognostic model. (A) OS analysis. Patients with OM-CMML had a significantly longer OS than did those with D-CMML (median OS, 131.81 vs 62.36 months; P = .038) and P-CMML (median OS, 131.81 vs 41.46; P = .001). (B) OS by Kaplan-Meier analysis of lower-risk patients by CPSS-P. Patients with OM-CMML did not have a significantly longer OS than did those with D-CMML (median OS: 131.81 vs 74.65 months; P = .199), but presented a significantly longer OS than did those with P-CMML (median OS, 131.81 vs 45.04; P = .005). (C) OS by Kaplan-Meier analysis of lower-risk patients by the Mayo prognostic model. Patients with OM-CMML had a significantly longer OS than did those with D-CMML (median OS, 131.81 vs 62.36 months; P = .046) and P-CMML (median OS, 131.81 vs 38.4; P < .001). OS was compared with 2-sided log-rank tests.
The predictive factors of evolution of OM-CMML into overt CMML are also independent adverse prognostic factors for survival in OM-CMML
At a median follow-up of 53.47 months, disease in 29.3% of patients with OM-CMML progressed to overt CMML. We assessed several variables to find predictors of a shorter time to CMML (ie, immunophenotypic profile, number of mutations, number of mutated genes, single gene mutations, RAS-pathway mutations, number of TET2 mutations, truncating vs nontruncating type TET2 mutations, and the prognostic scoring systems). Of them, we identified 3 variables with a significant capacity for predicting time to CMML: harboring >3 mutated genes (HR, 4.24; 95% CI, 1.076-16.711; P = .039), ASXL1 mutation (HR, 3.685; 95% CI, 1.034-13.131; P = .044), and having a PB monocyte percentage >20% (HR, 3.477; 95% CI, 1.054-11.471; P = .041). We observed a clear statistical trend when correlating ASXL1 mutation with the number of mutated genes (Spearman ρ, 0.31; P = .051). Then, we performed a Cox regression analysis including these 2 variables, and both retained their significance for predicting time to CMML (ASXL1; HR, 5.47; 95% CI, 1.35-22.18; P = .017; >3 mutated genes; HR, 6.03; 95% CI, 1.47-31.29; P = .014). Given the slightly better predictive capacity of harboring >3 mutated genes, this variable was selected instead of ASXL1 mutation for being introduced in the final multivariate model. This model included monocytes >20% and >3 mutated genes (monocytes, >20%: HR, 4.33; 95% CI, 1.23-15.20; P = .022; >3 mutated genes: HR, 5.82; 95% CI, 1.32-25.70; P = .02). Then, we implemented a score for predicting time to CMML: 0 points (none of them), 1 point (1 of them), or 2 points (both) that offered excellent accuracy (C-index, 0.73). Interestingly, these 2 variables were also independent adverse prognostic factors for OS in our OM-CMML series (monocytes: >20%; HR, 6.02; 95% CI, 1.66-21.84; P = .006; >3 mutated genes: HR, 7.63; 95% CI, 1.69-34.36; P = .008). These results are in line with previous observations of our group in which we found that patients with OM-CMML that evolved to CMML had a significantly shorter OS than those in whom the disease did not evolve.3 Of note, we consider that OM-CMML progressed to CMML when PB monocyte count was persistently maintained at >1 × 109/L. We detected 4 patterns in our series: (1) patients with OM-CMML having a PB monocyte count <1 × 109/L during all follow-up (n = 20; median follow-up, 44.1 months; IQR, 25.3-64.8); (2) a group with a monocyte count varying slightly above and under 1 × 109/L during follow-up with no definitive evidence of CMML progression (n = 8; median follow-up: 24.6 months; IQR, 8.5-68.7); (3) a group with a monocyte count varying slightly above and under 1 × 109/L at times, but with definitive evidence of CMML progression (n = 7; median time of evolution, 44.2 months; IQR, 34.3-60.4); and (4) a group of patients with a consistent monocyte count >1 × 109/L from the moment they exceeded the cutoff (n = 5; median time of evolution, 27.5 months; IQR, 14.8-48.7; supplemental Figure 1). Therefore, it is important not to assume a progression to CMML after a short follow-up, because approximately one-third of patients in our series displayed this oscillating behavior over a long period. This finding reinforces the idea of the continuum among these entities and could partially explain the divergent percentages of evolution of OM-CMML reported by different investigators.1,3,4
D-CMML evolution to P-CMML as a late evolutionary event in CMML
As mentioned, in our series, 29.3% of cases of OM-CMML evolved to overt CMML. This observation supports considering OM-CMML as an early stage of D-CMML and infers a continuum of OM-CMML, D-CMML, and P-CMML. In this sense, as previously reported by our group, the acquisition of second genetic hits such as RAS-pathway mutations, could partially promote the transition from one stage to another.3 Interestingly, at a median follow-up of 46.03 months, 28.6% of D-CMML in our series progressed to P-CMML. Remarkably, since the moment of progression to P-CMML, these patients had a very short OS (median OS, 10.61 months; 95% CI, 2.72-18.50). This finding reflects that P-CMML progression may be a late event in natural evolution of CMML.
In line with this finding, gene mutations associated with increased proliferation (ie, ASXL1 and RAS-pathway mutations)9,25-27 were identified as independent adverse prognostic factors in our series. We conducted univariate and multivariate analyses to evaluate the influence on OS of gene mutations observed in at least 10 patients (ie, ASXL1, CBL, RUNX1, SF3B1, SRSF2, TET2, and ZRSR2; Table 4). Moreover, we analyzed the prognostic impact of RAS-pathway mutations (ie, mutations in CBL, NRAS, and/or KRAS genes), observed in 22 of 94 patients (23.4%). Of those, the ASXL1, CBL, and RAS-pathway mutations had a significant impact on the univariate OS analysis (HR, 2.55; 95% CI, 1.18-5.51; P = .018; HR, 4.67; 95% CI, 1.87-11.69; P = .001; HR, 3.93; 95% CI, 1.78-8.67, P = .001; for ASXL1, CBL, and RAS pathway, respectively). Moreover, RUNX1 mutation showed a clear statistical trend for OS (HR, 2.71; 95% CI, 0.91-8.09; P = .075). Mutations in NRAS, TP53, and SH2B3 also showed an impact in univariate OS analysis (HR, 3.5; 95% CI, 1.17-10.49; P = .025; HR, 10.72; 95% CI, 3.79-30.29, P < .001; HR, 4.34; 95% CI, 1.47-12.81; P = .008; for NRAS, TP53, and SH2B3, respectively), but these were observed in a very small proportion of patients (7%, 5%, and 5%; for NRAS, TP53, and SH2B3, respectively) and were not included in subsequent multivariate analyses. In multivariate analyses for OS, which included ASXL1 and CBL mutations (HR, 2.26; 95% CI, 1.04-4.93; P = .04; HR, 4.16; 95% CI, 1.65-10.54; P = .003; for ASXL1 and CBL, respectively), or ASXL1 and RAS-pathway mutations (HR, 2.47; 95% CI, 1.13-5.37; P = .023; HR, 3.91; 95% CI, 1.74-8.77, P = .001; for ASXL1 and RAS pathway, respectively), ASXL1, CBL, and RAS-pathway mutations retained their significance (Table 5).
Discussion
Patients with OM-CMML are currently categorized in the various MDS categories of the 2017 WHO classification,15,16 but they display clinical, morphological, cytogenetic, molecular, and immunophenotypic profiles similar to those of patients with overt CMML.1,3,4 By definition, these patients present a relative monocytosis in the absence of absolute monocytosis and leukocytosis. The proliferative phenotype of CMML (ie, the presence of at least 13 × 109/L leukocytes) has been associated with a mutational profile enriched in the RAS pathway,25-27 ASXL1,9 SETBP1,9,12 and JAK228 mutations. JAK2 mutations are observed in only ∼5% to 10% of patients with CMML, they are usually subclonal events, and the progressive increment in their VAFs are typically associated with leukocytosis and thrombocytosis.28 Different from the rest of the gene mutations associated with leukocytosis, JAK2 mutations have not been associated with worse outcomes in this disease.28 It may be very difficult to distinguish OM-CMML or CMML from myeloproliferative neoplasms with relative and/or absolute monocytosis (eg, essential thrombocythemia with monocytosis, prefibrotic primary myelofibrosis with monocytosis, and primary myelofibrosis with monocytosis). For these cases, the only way to establish an accurate diagnosis is to perform a BM biopsy with a detailed histologic examination. In this scenario, the analysis by FC of the distribution of peripheral blood monocyte subsets seems to be a very useful tool for discriminating between myeloproliferative neoplasms with monocytosis and CMML.29,30 Differential diagnosis with other situations such as systemic mastocytosis with an associated CMML requires further diagnostic workup (eg, KIT mutation analysis, FC assessment of expression of CD2 and CD25 on mast cells, blue toluidine staining of the BM) in addition to a histological examination of a BM biopsy specimen.
As previously reported by our group, we found a positive correlation between mutations in the RAS-pathway and ASXL1, depicting a partial interplay among ASXL1, RAS-pathway mutations, and the proliferative phenotype of CMML: 3 well-accepted independent adverse prognostic factors in this disease.3 Currently, ASXL1, RUNX1, SETBP1, and NRAS mutations have been detected as those with the best outcome prediction accuracy in CMML, and for this reason, they have been selected to implement the CPSS-mol.12 As previously reported, RAS-pathway mutations are very rare in patients with OM-CMML1,3,4 and if present, these are more likely to constitute small clones.4 Moreover, according to the current definition,1 patients with OM-CMML must present <10 × 109/L leukocytes; if not, they would meet CMML diagnostic criteria. Therefore, it could be expected that OM-CMML, representing the first step within the proliferative continuum of CMML, would have better outcomes than the more advanced stages, D-CMML and P-CMML. In a recent work published by Montalban-Bravo et al,4 patients with OM-CMML had a significantly better OS than P-CMML and presented a clear statistical trend when compared with D-CMML. Moreover, they observed a better OS when comparing OM-CMML with overt CMML in the univariate survival analysis, but OM-CMML was not confirmed as an independent favorable prognostic factor when adjusted by the CPSS and age. They argued that this finding, together with the clinical-biological similarities of both entities, reinforced the consideration of OM-CMML as a subtype of CMML. In our view, the differences in prognosis between OM-CMML and overt CMML do not invalidate the consideration of OM-CMML as a specific subtype of CMML.
In recent years, we have witnessed an exponential increase in genomic knowledge of the disease, which has enabled us to partially understand the prognostic differences between the dysplastic and proliferative subtypes of CMML. With a median follow-up ranging from 12 to 31 months in the different series analyzing the clinicopathological features of OM-CMML, a ∼20% to ∼40% of progression to D-CMML was described.1,3,4 This fact and the current genomic knowledge suggests that OM-CMML is an early step of D-CMML.1,3 In this sense, as previously reported by our group, the acquisition of second genetic hits, such as RAS-pathway mutations, could partially promote the transition from one stage to another.3 Hence, it would not be surprising that, as a group, patients with OM-CMML present better outcomes than do patients with D-CMML. To reinforce the continuum among OM-CMML, D-CMML, and P-CMML, it would also be interesting to demonstrate whether a proportion of patients with D-CMML will progress to P-CMML during disease evolution, reinforcing the idea of a proliferative continuum in CMML, with biological and prognostic implications.
Our work has helped us to explore some of these open questions. First, patients with OM-CMML had a significantly longer OS and LFS than did those with D-CMML and P-CMML. Furthermore, OM-CMML was recognized as an independent favorable prognostic factor in our CMML series adjusted by age, CPSS, CPSS-P, and the Mayo prognostic model. Moreover, by adding OM-CMML we observed an improvement in the predictive power for OS of these 3 scores. In the same line, gene mutations associated with increased proliferation (ie, ASXL1 and RAS-pathway) were identified as independent adverse prognostic factors in our series.
Second, at a median follow-up of 53.47 months, 29.3% of patients with OM-CMML progressed to overt CMML. The different groups dedicated to the study of OM-CMML have reported different data on the proportion of patients in whom the disease progresses to CMML (ie, Montalban-Bravo et al: 40%, median follow-up, 13.5 months4 ; Geyer et al: 38%, median follow-up, 12 months1 ; Calvo et al: 18%, median follow-up: 31.1 months3 ). As previously described, we observed 4 different patterns of evolution. Two of these patterns were characterized by a monocyte count varying slightly above and under 1 × 109/L monocytes during follow-up; one of them with no definitive evidence of CMML evolution, and the other finally progressing to overt CMML. Probably, all patients with OM-CMML who have this oscillating behavior would be better considered together in a single group, because those who finally had disease progression, had a longer median follow-up than those who did not (44.2 vs 24.6 months). Therefore, it is important not to assume a progression to overt CMML after a short follow-up. This finding, in addition to reinforcing the idea of the biological continuum between OM-CMML and overt CMML, enables us to understand partially the different percentages of progression referred by the different authors.1,3,4 In future studies with larger series of patients, it would be interesting to investigate the main biological differences among patients included in these 3 different groups with different patterns of evolution, to provide a new layer of understanding in the evolutionary continuum of these entities. As previously reported by our group, patients with OM-CMML that evolved to CMML had a significantly shorter OS.3 Therefore, it seemed interesting to find predictors of evolution of OM-CMML into CMML. In this sense, we identified 3 variables with a significant capacity for predicting time to CMML: harboring more than 3 mutated genes, ASXL1 mutations, and a PB monocyte percentage >20%. These variables were also identified as independent prognostic factors for OS in OM-CMML.
Third, at a median follow-up of 46.03 months, 28.6% of patients with D-CMML evolved to P-CMML. To the extent of our knowledge, this is the first time that it has been offered as a formal demonstration that, at least in some cases, P-CMML represents an evolutionary step of D-CMML. In our series, D-CMML that progressed to P-CMML displayed a median OS of only 10.61 months since the moment of evolution. This finding reflects that the evolution to proliferative type of the disease may be a late event in the natural history of CMML.
In summary, OM-CMML had better outcomes than D-CMML and P-CMML. The evolution of a high proportion of patients with OM-CMML into D-CMML, and a similar percentage of patients with D-CMML into P-CMML reinforces the idea of the proliferative continuum of CMML. The clinical outcomes of OM-CMML, partially explained by its genomic profile, support its consideration as the first stage in the evolutionary spectrum of CMML.
Acknowledgments
The authors thank the Parc de Salut Mar Clinical Research Ethics Committee of the Hospital del Mar Research Institute for study approval. This study was supported in part by grants from ISCI II-FEDER FIS PI16/0153, FIS PI17/313, FIS PI19/0005, 2017SGR205, and 2017SGR437, and Xarxa de Banc de Tumors de Catalunya.
Authorship
Contribution: X.C. designed the study; all authors collected and assembled data from the study patients; X.C., D.R.-B., N.G.-G., J.G., and L.A. analyzed the data; X.C., D.R.-B., N.G.-G., J.J.R.-S., and L.A. interpreted the data; X.C. wrote the manuscript; and all authors reviewed and approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xavier Calvo, Hospital del Mar, Paseo Marítimo, 25, 08003 Barcelona, Spain; e-mail: xcalvo@psmar.cat.



