Key Points
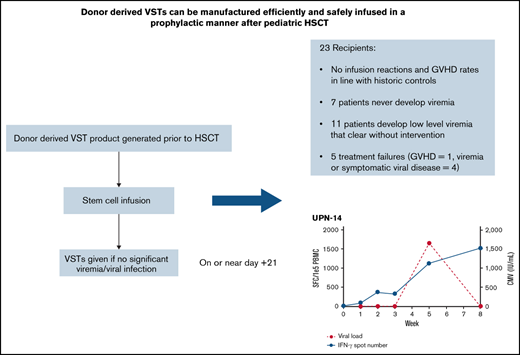
Donor-derived VSTs can be manufactured and administered early posttransplant in a prophylactic manner without increased risk of GVHD.
Scheduled infusion of VSTs is associated with few treatment failures and a seeming ability to clear low-level viremia.

Abstract
Infections with double-stranded DNA viruses are a significant cause of morbidity and mortality in pediatric patients following allogeneic hematopoietic stem cell transplantation (HSCT). Virus-specific T-cell therapies (VSTs) have been shown to be an effective treatment for infections with adenovirus, BK virus, cytomegalovirus (CMV), and Epstein-Barr virus (EBV). To date, prophylactic regimens to prevent or mitigate these infections using conventional antiviral medications provide suboptimal response rates. Here we report on a clinical trial (NCT03883906) performed to assess the feasibility of rapid manufacturing and early infusion of quadrivalent VSTs generated from stem cell donors (“donor-derived VSTs”) into allogeneic HSCT recipients with minimal or absent viremia. Patients were eligible to receive scheduled VSTs as early as 21 days after stem cell infusion. Twenty-three patients received scheduled VSTs. Twenty of 23 patients had no viremia at the time of infusion, while 3 patients had very low-level BK viremia. Two developed clinically significant graft-versus-host disease (GVHD), although this incidence was not outside of expected incidence early after HSCT, and both were successfully treated with systemic corticosteroids (n = 2). Five patients were deemed treatment failures. Three developed subsequent significant viremia/viral disease (n = 3). Eighteen patients did not fail treatment, 7 of whom did not develop any viremia, while 11 developed low-level, self-limited viremia that resolved without further intervention. No infusion reactions occurred. In conclusion, scheduled VSTs appear to be safe and potentially effective at limiting serious complications from viral infections after allogeneic transplantation. A randomized study comparing this scheduled approach to the use of VSTs to treat active viremia is ongoing.
Introduction
T lymphocytes are required for the control and eradication of viral infections.1 Infections with double-stranded DNA (dsDNA) viruses like cytomegalovirus (CMV), Epstein-Barr virus (EBV), adenovirus (AdV), and BK virus (BKV) are a significant source of morbidity and mortality following allogeneic hematopoietic stem cell transplantation (HSCT) due to the prolonged periods of myelosuppression and immunosuppression needed to promote engraftment.2-5 Our own institutional prevalence of infection with these viruses by transplant day +100 ranges from 20% to 54% depending on the virus.6 The number of concurrent dsDNA viral infections and the viral load area under the curve are significant risk factors for early and late mortality after HSCT.7 However, both prevention and treatment of viral infections can be difficult. There are currently no US Food and Drug Administration (FDA)-approved therapies for the prevention and treatment of AdV and BKV, and agents like ganciclovir, foscarnet, and rituximab used for the management of CMV and EBV have high rates of organ toxicity and suboptimal response rates, while often prolonging hospitalizations.8-12
Virus-specific T cells derived from a patient’s stem cell donor (donor-derived VSTs) can be rapidly manufactured using pools of overlapping viral antigenic peptides and safely infused for the preemptive treatment of the aforementioned viruses.13,14 This approach has been effective without an increase in the development of de novo graft-versus-host disease (GVHD).15-17 Both autologous and allogeneic VSTs have also been used for prophylaxis against posttransplant lymphoproliferative disease, but data on the use of VSTs for prevention or mitigation of viremia are lacking.18 Here, we present data from a single-arm, phase 2 trial using scheduled VSTs on or near posttransplant day +21 in patients with no more than clinically insignificant degrees of viremia and no evidence of invasive viral infection as prophylaxis against significant viremia or invasive viral disease.
Methods
Study population and clinical trial
This single-arm, phase 2 study was approved by the Cincinnati Children’s Hospital Medical Center (CCHMC) Institutional Review Board and cleared by the FDA (#NCT03883906). All allogeneic HSCT recipients at CCHMC were eligible but required separate and prior enrollment of both the recipient and donor on a separate study allowing for the generation of donor-derived VST (NCT02048332). Patients meeting eligibility criteria and completing consent were able to receive VST products no sooner than 21 days after stem cell infusion. Patients received 2 × 107 VST/m2 as a single infusion. Eligibility for prophylactic VST infusion required blood AdV polymerase chain reaction (PCR) <1000 copies/mL, CMV PCR <500 IU/mL, EBV PCR <9000 IU/mL, BKV PCR <1000 copies/mL, no evidence of invasive CMV or AdV infection, no evidence of EBV-associated lymphoproliferation, and no evidence of symptomatic BKV such as hemorrhagic cystitis. Acyclovir at prophylactic dosing for the prevention of herpes simplex virus and varicella-zoster virus was allowed. Clinical status had to allow for tapering of any ongoing steroids to ≤0.5 mg/kg of prednisone equivalents. Patients were required to be at least 2 weeks removed from their last dose of alemtuzumab with a quantified alemtuzumab level of <0.15 µg/mL.19 Exclusion criteria included viral infection or reactivation defined by not meeting the infectious criteria previously mentioned, active acute grade 2 to 4 GVHD, infusion of antithymocyte globulin (ATG) within 2 weeks of infusion, and uncontrolled relapse of malignancy. The primary endpoint of the study was to establish the feasibility of producing VSTs and safely infusing as early as 21 days following stem cell transplant without excessive infusional toxicity or an increased incidence of acute GVHD (aGVHD). The study would be feasible if a VST product was successfully manufactured for >75% of patients. Excessive infusional toxicity was defined as having 5 attributable grade 3 to 4 infusional toxicity events in the first 27 patients infused. The historical norms of grade 2 to 4 GVHD at our institution is 15%, and the study was initially generated with a Simon 2-stage design, with acceptable rates of grade 2 to 4 GVHD being <29% in the first stage and <23% in the second stage. The secondary endpoint was clinical efficacy (as defined in the “Response criteria” section below). All infused patients were followed through transplant day +100.
VST manufacturing
VSTs targeting AdV, BKV, CMV, and EBV were generated as previously described.20 Products were required to meet all release criteria for safety, sterility, and alloreactivity prior to infusion. The percentage of T cells secreting interferon-γ in response to pooled viral antigens was performed as previously described.20
Response criteria
We anticipated that even with the infusion of scheduled VSTs, numerous patients would still develop modest degrees of viremia, which would then trigger expansion of VSTs followed by suppression of viremia without the need for antiviral agents. We, therefore, expected a reduction in peak levels of viremia and the need for antiviral therapy. Thus, thresholds for determining failure of treatment were chosen to be clinically and statistically meaningful and represent levels at which patients would benefit from other preemptive antiviral therapies. Failure cutoffs were chosen by consensus agreement of senior transplant physicians. The failure cutoffs used in this study, when applied to a prior institutional cohort of 113 consecutive recipients of donor-derived VSTs, resulted in a treatment failure rate from viremia of 17%.
Patients were considered treatment failures if any of the following occurred prior to day +100 after transplant: blood AdV PCR >50 000 copies/mL, BKV PCR >100 000 copies/mL, CMV PCR >5000 IU/mL, EBV PCR >100 000 copies/mL, evidence of invasive AdV or CMV infection, evidence of EBV-associated lymphoproliferation, evidence of symptomatic hemorrhagic cystitis, initiation of new antiviral therapy (medication and/or subsequent VST product), or the development of grade 3 to 4 aGVHD. Surveillance PCR testing for all 4 viruses was checked at least weekly for the first 30 days after infusion and then generally weekly but no less than monthly through transplant day +100. aGVHD diagnosis, with stage and grade per Glucksberg criteria, were determined by the treating physician in realtime through transplant day +100 and reviewed and confirmed by senior transplant physicians at day 100, and when available, confirmed by biopsy specimens.21
Enzyme-linked immunosorbent assay (ELISpot)
Peripheral blood mononuclear cells (PBMCs) were isolated from the peripheral blood (PB) of VST recipients, with samples collected immediately prior to the VST infusion, weekly for the first month after infusion of VSTs, and then monthly through transplant day +100. Interferon-γ ELISpot was then performed using these samples to assess for the presence and proliferation of antiviral T cells in the PB over time as previously described.20
Results
Patient characteristics
The study period began in March 2019, with the last enrollment in October 2020. During this time, 138 patients at CCHMC underwent their first allogeneic HSCT. Of those, 30 (21.7%) enrolled in the study. The most common reason for nonenrollment was a lack of VST product, which occurred in 44 patients (31.9%). Reasons to not have product included donor declining blood draw, donor and/or recipient residing in a country that did not allow procurement of research samples, manufacturing failure, recipient of cord blood graft, and delays in donor sample collection for VST manufacturing leading to delays in product generation. Other reasons for nonenrollment included disqualifying levels of viremia/viral infection (these patients were eligible for treatment infusion of VST; 23/138 [16.7%]), primary physician preference due to clinical instability or other cause (16/138 [10.6%]), patient/family decline (12/138 [8.7%]), and primary graft failure (3/138 [2.2%]). One haploidentical HSCT recipient received prophylactic VSTs and developed skin GVHD requiring treatment with systemic steroids, and as a result, all recipients of haploidentical transplant were subsequently excluded from the study to remove significant HLA mismatch as a potential confounding risk factor and to allow for greater cohort homogeneity. During the study period, 10/138 (7.2%) patients undergoing their first allogeneic HSCT were recipients of haploidentical transplantation and were thus ineligible.
Of the 30 patients who enrolled, 23 ultimately received prophylactic infusions. Reasons for not receiving infusions were acute medical decompensation prior to VST infusion (n = 2), development of disqualifying levels of viremia between consent and infusion (n = 3; all subsequently received donor-derived VSTs on a preemptive treatment protocol), and patient/family withdrawal prior to infusion (n = 2). Demographic information on the 23 infused patients is shown in Table 1; 20/23 had no viremia at the time of infusion, while 3 patients had <1000 copies of BKV. Of the 5 patients who received alemtuzumab prior to HSCT, the median alemtuzumab level prior to VST infusion was 0.05 µg/mL (range, 0-0.12). One patient was receiving corticosteroids (for immunosuppressive purposes) at the time of infusion at a dose of 0.37 mg/kg per day of prednisone. No patients had a prior history of GVHD.
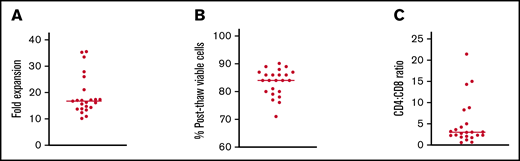
VST product characteristics
All patients received VST products manufactured from their stem cell donor. VSTs were generated from donors fully matched with their recipients in 20 cases and mismatched at 1 HLA allele in 2 cases. One patient received a haploidentical graft that matched at 8/10 HLA alleles. EBV and CMV serologies were known for all donors and showed prior infection in 12/23 (52.2%) and 18/23 (78.3%) of donors, respectively. T cells with activity against EBV and CMV were not able to be generated from donors who were seronegative for those specific viruses. Of the 13 patients who had donors that were seronegative for EBV and/or CMV, 7 were seropositive for the respective virus pretransplant, while 6 were seronegative. Donor BKV and AdV serologies were not tested due to the ubiquitous nature of these viruses. The fold expansion of cells in culture was 16.70-fold (range, 10.1-35.5) with a median postthaw viability of 84% (range, 71-90%) (Figure 1A-B). CD4+ cells outnumbered CD8+ cells in 21/23 (91.3%) products. The median CD4+:CD8+ ratio was 3.0 (range, 0.7-21.4) (Figure 1C). The median percentage of intracellular interferon–γ-positive T cells by flow cytometry was 0.33% for CMV (range, 0.00-10.11), 3.98% for ADV (range, 1.43-14.91), 0.21% for BKV (range, 0.00-5.78), and 0.10% for EBV (range, 0-3.6). Among seropositive donors, the median for CMV was 2.29% (range, 0.02-10.11), and for EBV was 0.24% (range, 0.00-3.6).
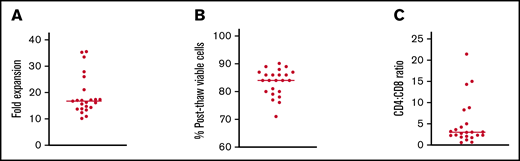
Preclinical testing of VST products infused into patients on this study. (A) Fold expansion of VSTs in culture. (B) Percentage of viable cells after thawing of cryopreserved products. (C) Ratio of CD4:CD8 T cells in each product. Lines in all panels represent median values.
Preclinical testing of VST products infused into patients on this study. (A) Fold expansion of VSTs in culture. (B) Percentage of viable cells after thawing of cryopreserved products. (C) Ratio of CD4:CD8 T cells in each product. Lines in all panels represent median values.
Prophylactic VST administration early after HSCT is feasible with an acceptable safety and tolerability profile
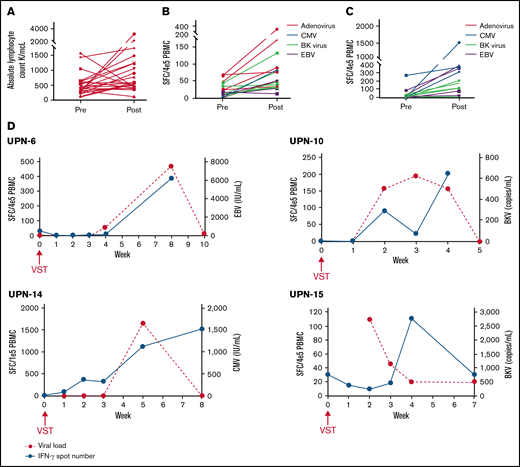
Of 138 patients, 116 (84.1%) enrolled in the study for the generation of donor-derived VSTs. Only 11/116 (9.5%) of those patients and 11/138 (8.0%) of the entire cohort did not have a product available due to manufacturing issues. An additional 4 patients had VSTs available but not until after day +21, leading to a total of 15/138 (10.9%) patients who did not have cells available by this early time point, although this study did allow enrollment of patients with delayed VSTs. Of note, 15 additional patients had products that met all safety and release criteria and thus were available for preemptive treatment of viremia but not acceptable for this study due to manufacturing during a 3-month audit of laboratory procedures. These products were not considered manufacturing failures. As >75% of patients had a product manufactured successfully, the feasibility endpoint was met. Between the 11 patients who did not have a product available due to manufacturing and the 23 patients excluded due to early viral reactivation, a total of 34/138 (24.6%) patients were unable to enroll due to objective predefined criteria. As a result, 75.4% of patients were considered for inclusion. The median day of infusion was transplant day +23 (range, day +21 to +40). The median absolute lymphocyte count (ALC) at infusion was 380 (range, 130-1550) (Figure 2A). Eleven of 23 patients received VSTs on the first day of eligibility. All patients received the target cell dose of 2 × 107 VST/m2. The most common reasons for delay were due to scheduling/logistics of infusion (n = 7), delay in consent process (n = 2), delay in VST generation (n = 2), primary physician preference (n = 1), and awaiting alemtuzumab clearance (n = 1). Only 3 infusions occurred on or after day +30: 1 due to manufacturing delays (infused on day +30), 1 due to primary physician preference, and 1 due to awaiting alemtuzumab clearance. The median ALC 30 days after infusion was 560 (range, 120-3210); ALC was increased in 17/23 (73.9%) patients (Figure 2A). Two of 6 (33.3%) patients with decreased ALC received systemic corticosteroids within the first 30 days after VST infusion, which may explain the lack of lymphocyte expansion.
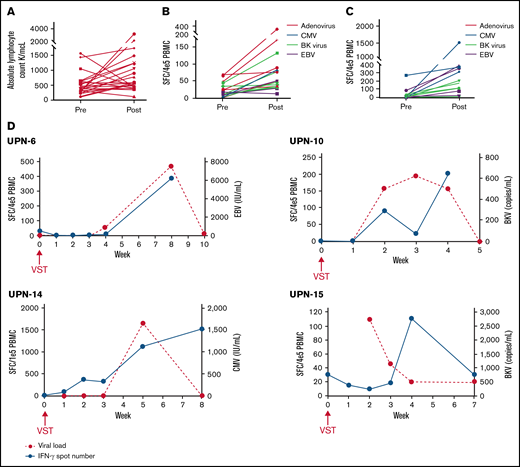
Increase in antiviral T cells in PB of patients after receiving scheduled VSTs. (A) Absolute lymphocyte counts at infusion (pre) and 30 days after VST infusion (post) in all recipients. (B,C) Baseline (preinfusion) and peak (any point postinfusion) quantitated antiviral T cells as determined by interferon-γ ELISpot in recipients without any viremia and with viremia that cleared without intervention, respectively. (D) Representative examples of ELISpots from nontreatment failure patients with viremia that resolved with corresponding curves showing the kinetics of viremia.
Increase in antiviral T cells in PB of patients after receiving scheduled VSTs. (A) Absolute lymphocyte counts at infusion (pre) and 30 days after VST infusion (post) in all recipients. (B,C) Baseline (preinfusion) and peak (any point postinfusion) quantitated antiviral T cells as determined by interferon-γ ELISpot in recipients without any viremia and with viremia that cleared without intervention, respectively. (D) Representative examples of ELISpots from nontreatment failure patients with viremia that resolved with corresponding curves showing the kinetics of viremia.
Scheduled VSTs were given early in the posttransplant period. As a result, we anticipated seeing some GVHD in our cohort but did not expect the incidence to be above our historical norm. In a cohort of 200 consecutive allogeneic transplant recipients at our institution between 2015 and 2018, the rate of grade 2 to 4 GVHD within 100 days of transplant was 14.5%, and the rate of grade 3 to 4 GVHD was 7%. Two cases of GVHD occurred within 30 days of VST infusion and were considered possibly attributable to VSTs: 1 patient with grade 2 skin GVHD and 1 patient with grade 3 to 4 skin and gastrointestinal (GI) GVHD; GVHD developed on transplant day +40 (14 days after VST infusion) and day +38 (17 days after VST infusion), respectively.
Three additional patients developed GVHD >30 days after infusion, all low grade (grade 1 skin only in 2 patients, grade 1 skin and grade 2 GI in 1 patient) and were considered not attributable to VSTs. The incidence of grade 2 to 4 and 3 to 4 GVHD by transplant day +100 in this cohort was 3/23 (13.0%) and 1/23 (4.3%), which are in line with our prior experience of the incidence of GVHD in this population. The study was initially designed to infuse 122 patients, and the prespecified safety endpoint was ≤28 patients (23.0%) with grade 2 to 4 GVHD at any point following infusion. Since only 13% of patients in this initial cohort had grade 2 to 4 GVHD, we opted to close this study and proceed with a successor randomized study comparing scheduled VSTs with preemptive VSTs, which is now ongoing.
There were no cases of cytokine release syndrome. Transplant-associated thrombotic microangiopathy was seen in 2 cases (8.7%) diagnosed at 6 weeks and 18 months after VSTs, respectively. Both were treated with eculizumab, and no infusion reactions occurred.
Treatment failure occurs in a minority of patients infused with prophylactic VSTs
Five of the 23 patients infused with VSTs were classified as treatment failures (21.7%). Patients who failed treatment are described in Table 2. One patient failed due to the development of EBV viremia requiring treatment with rituximab that arose after receiving steroids for the management of grade 2 skin GVHD. This patient was the recipient of a haploidentical transplant and haploidentical VSTs. One patient failed due to the development of grade 3 to 4 skin and gut GVHD occurring within 30 days of infusion; this patient was also treated with steroids and subsequently also developed significant ADV viremia and mild EBV viremia. Two patients were treatment failures due to peak viremia levels meeting the protocol failure threshold, 1 for CMV and 1 for EBV (peak levels of 9400 and 528 917, respectively). In both cases, patients cleared the viremia with subsequent antiviral therapy (valganciclovir for CMV and rituximab followed by third-party VSTs for EBV). The final treatment failure had symptomatic BKV dysuria; this did not require further antiviral therapy and ultimately cleared, but the patient did require admission for pain control. Of the 3 patients who failed therapy due to either CMV or EBV viremia, donor serology was positive for the respective virus in all 3 cases. Of the 4 patients who were treatment failures due to viremia or viral disease, 2 patients (#1 and #23) had VST products with antiviral activity greater than the median value for that virus within the entire cohort, while 2 patients (#9 and #20) had products with activity that was lower than the median.
Four treated patients died, all after day +100. Two of these deaths were in patients who were treatment failures: 1 of bacterial infection 18 months postinfusion and 1 from multiorgan failure in the context of GVHD and bacteremia at 5.5 months postinfusion. Two were in patients who were not treatment failures, both of whom died of relapse of leukemia (4 months and 15 months postinfusion, respectively). The median posttransplant day at death was day +347 (range, 143-675). All other patients are currently alive, although 2 have had a relapse of malignancy at 6 and 10 months postinfusion, respectively.
Eleven of 18 of the nonfailure patients developed viremia from at least 1 virus (Table 3). Three patients had low-level CMV viremia (median peak viremia of 1647; range, 1112-2329), which in all cases resolved without additional intervention. Five patients had low-level BKV viremia (median peak viremia of 623; range, 500-5513), which in all cases resolved without intervention. Six patients had low-level EBV viremia (median 4564; range, 500-46 751). None of these patients required EBV-directed therapy, and viremia resolved entirely in 2 patients while there was residual viremia of 9206 in 2 patients and <200 in 3 patients at day +100, which then subsequently cleared without intervention. Donor serology was positive for the respective virus in the 8 patients who had any EBV or CMV viremia (2 patients with CMV, 5 patients with EBV, 1 patient with both). Seven of the 18 nontreatment failures (38.9%) did not develop any viremia by transplant day +100 (Tables 3 and 4).
ATG was used during conditioning in 3/5 (60.0%) treatment failures but was used in only 3/18 (16.7%) nonfailing patients. Alemtuzumab was given in conditioning to 1/5 (20.0%) treatment failures and in 4/18 (22.2%) nonfailing patients, but unlike in the ATG setting, antibody clearance was monitored and ensured in these cases. Abatacept was given after transplantation to 2/5 (40.0%) of treatment failures and 6/18 (33.3%) of nonfailing patients.
Antiviral T cells are present in PB of VST recipients and poised to expand upon detection of viral ligand
Interferon-γ ELISpot assays were performed for all VST recipients on weekly samples of PBMCs drawn for the first month and then monthly thereafter when possible.
Of the 7 patients who never developed viremia, increased viral directed T cells were detected in the PB in 6 patients, starting from a baseline median of 14 spot-forming cells (SFC)/4 × 105 PBMC (range, 1.4-69) to a median peak of 49.8 SFC/4 × 105 PBMC (range, 29-315) (Figure 2B). An increase above the baseline was seen in 16/17 (94.1%) of the ELISpot assays performed in this cohort. One patient, who had a donor who was EBV- and CMV-negative, was an outlier with very high baseline T-cell numbers against ADV and BKV (>500) that subsequently decreased to 0 at all other time points and was excluded from analysis due to concern for an inaccurate baseline study.
In the 11 patients with 15 distinct viremias that improved without intervention, an increase in viral directed T-cell count above the baseline was seen on ELISpot in all 15 instances, going from a median baseline of 7.0 SFC/4 × 105 PBMC (range, 0-271.0) to a median peak of 177.0 SFC/4 × 105 PBMC (range, 8.0-1517.0) (Figure 2C). Representative examples showing the kinetics of viral clearance in association with a corresponding increase in antiviral T cells are shown in Figure 2D.
Two treatment failures (1 patient with CMV viremia that resolved after adding valganciclovir and 1 patient with symptomatic BK cystitis that subsequently resolved without intervention but required hospitalization for pain control) had increases in spot number by ELISpot with peaks of 276.2 and 14.0 SFC/4 × 105 PBMC, respectively. The sample size is small, but there were no obvious qualitative differences in the kinetics of T-cell expansion or elevation in these patients compared with nonfailure patients. ELISpots from the 3 other treatment failures had confounding factors that make interpretation difficult; in 2 cases, the patients received lympholytic steroids, and in 1 case, the viremia started after the research samples had been collected.
Discussion
In this feasibility study, we report the prophylactic administration of scheduled quadrivalent donor-derived VSTs to 23 pediatric patients with no or very minimal viremia early after allogeneic HSCT. Most patients either never developed viremia or developed low-level viremia that cleared without subsequent viral directed therapy. VSTs were infused at a median transplant day +23, a time during which there are few or no endogenous functional T cells. We believe our data support the hypothesis that scheduled VSTs from mostly fully HLA-matched stem cell donors can persist in recipients, poised to expand upon detection of viral ligand. Accordingly, this approach may allow for decreased complications from dsDNA viruses after HSCT.
Efficacy outcomes on this study were promising, although this was not the primary endpoint and needs to be cautiously interpreted in this single-arm study. More definitive efficacy conclusions require a randomized study which is now ongoing. In this study, 7 patients (30.4%) never developed viremia by day +100 after transplant. It is notable that all 7 of these patients had a 10/10 HLA match with their donors, and 4/7 received grafts from siblings, so this is generally a population at lower risk for viral infection after HSCT.22,23 There were 11 patients who did develop low-level viremia but were able to clear the infection without further intervention. Interestingly, in each case of viremia, there was a corresponding increase in antiviral T cells on ELISpot following the initial detection of viremia by PCR. As this occurred early after transplant while the ALC is low, we presume that this increase in interferon–γ-secreting T cells is primarily made up of the donor-derived VSTs, although we did not definitively demonstrate this. These data suggest that donor-derived VST infused in the absence of viral ligand persists and can expand upon encountering viral antigen (Figure 2D). T-cell receptor sequencing could further demonstrate whether T cells arose from VSTs or the stem cell graft,24 and this will be an area of future study.
The goal of this study was to infuse as close to 21 days after stem cell infusion as possible, as generally, this is around or before most viremias occur.6,25,26 A total of 44 patients (31.9%) of the cohort were not eligible due to lack of VST product; however, 11/44 were due to manufacturing failures, and 15/44 were due to otherwise acceptable products not used during a laboratory audit. As a result, only 18/138 (13.0%) lacked a product for reasons outside of our control. With expected decreases in manufacturing failures over time, we believe the number of patients who will not have a product will be acceptably low, although this will be answered on an ongoing successor randomized study. Despite infusing T-cell products at a time when aGVHD often first develops, only 2 patients developed GVHD during the first 30 days after infusion, and only 1 was grade 3 to 4.27,28 Importantly, the historical rates of grade 2 to 4 and grade 3 to 4 GVHD at our institution in a cohort of 200 consecutive patients was 14.5% and 7%, respectively, making the incidence of GVHD seen in this study in line with prior institutional norms. Of note, the protocol was amended to make recipients of haploidentical transplants ineligible due to GVHD in 1 recipient. Haploidentical transplants make up a small percentage of HSCT at our institution but are increasing throughout the field as a whole. While our successor trial excludes patients with a match <9/10, to definitively determine the risk to recipients of haploidentical transplant, we plan to open a separate study for solely those patients with stringent stopping rules surrounding the development of GVHD.
VST therapy, whether from an individual’s stem cell donor or a third-party donor, has been shown to be a safe and effective approach for the preemptive treatment of these viral infections, even in patients who have failed conventional antiviral therapy.16,29-33 VSTs have largely been used in the treatment rather than the prevention of viral infections. Medical prophylaxis regimens are incompletely effective at preventing viral reactivation and often result in undesired toxicities. Letermovir has shown promise for prophylaxis in CMV-positive patients, although it is currently only FDA approved in adults.34 As a result, trialing VST for prevention is rational. One small study of 12 recipients showed VSTs could be safely infused as early as transplant day +2 (with a median for their cohort of day +13) with a tolerable side effect profile.35 CMV reactivation was seen in 50% of patients in this study, although all were in the context of steroid therapy. Another recent study demonstrated prophylactic delivery of VSTs to 11 adult patients but at a median infusion time of day +37 (range of day +28 to +76).36 Additional antiviral treatment was only required in patients who received steroids, although 4 patients did develop grade 3 to 4 GVHD. The data from this study support our hypothesis that scheduled donor-derived VSTs limit viral infections and obviate the need for antiviral medications after HSCT. Both prior studies enrolled only adult patients, and to our knowledge, our cohort represents the first study investigating prophylactic VSTs in pediatric patients. We believe this study supports the use of scheduled VSTs, not necessarily to decrease the percentage of patients who develop viremia but instead to limit the degree of viremia and the need for conventional antiviral medications. Currently, we have an ongoing randomized study comparing the prophylactic approach to the use of VSTs for the treatment of viremia, and if confirmed, this approach could become a new standard of care for viral prophylaxis. If pediatric clinical trials of letermovir show similar outcomes as adult studies, adding letermovir in combination with scheduled VSTs would be testable and reasonable.
This open-label study was limited by the lack of a control arm. However, it is promising that only 3/23 (13.0%) patients received additional antiviral therapy. Although it is difficult to make direct statistical comparisons with a historical cohort due to possible selection biases, it is notable that in a cohort of 200 consecutive allogeneic transplant recipients at our institution from the immediate pre-VST implementation period, 46% of patients required treatment with conventional antiviral therapy.
A limitation of this study is the small sample size, with only 23 patients receiving a scheduled VST product, accounting for 16.7% of all allogeneic transplant recipients during the study period and 19.8% of patients enrolled on the protocol to generate donor-derived VSTs. It is reassuring that only 8.0% of patients did not receive an infusion due to a lack of available product, suggesting enrollment can be optimized on successor studies through changes in eligibility criteria. The small sample size was in part driven by excluding patients with ongoing viremia or viral infection. Our ongoing successor randomized trial will attempt to overcome this confounding factor by including patients with preexisting viremia to determine if VSTs given at a scheduled early time point of day +21 have improvements with regard to viral complications and peak viremia as compared with the standard of care strategy of receiving treatment VSTs at the provider’s discretion. It is also likely that there will always be a proportion of recipients without donor-derived VSTs. Third-party VSTs are an important option in these cases, at least for treatment, if not prophylaxis. One additional weakness is that since the VSTs are not tagged, we cannot definitively show that expanded populations of T cells originate from the VST. Since VSTs are given at a time of profound lymphopenia, we presume the expansion seen on ELISpot is of donor origin, but future studies will incorporate T-cell receptor sequencing to further establish this point.
In summary, the administration of scheduled VSTs as a preventative measure in patients with no or minimal viremia was safe with GVHD rates in line with our institutional standard. These data, if supported by additional studies, raise the prospect of an important change in the standard of transplant care, with most recipients likely to benefit from this strategy.
Acknowledgments
The authors thank the staff of the Regenerative Medicine and Cellular Therapies Division at Hoxworth Blood Center for their critical aid in managing and manufacturing VSTs and infusions and the CCHMC Bone Marrow Tissue Repository and the Cell Processing Core laboratories for ongoing technical assistance. This study was supported by a St. Baldrick’s Fellow award (J.D.R.) as well as divisional funds from the Division of Bone Marrow Transplant and Immune Deficiency in the Cancer and Blood Diseases Institute of Cincinnati Children’s Hospital Medical Center.
Authorship
Contribution: J.D.R., S.M.D., A.S.N., and M.S.G. designed the study, performed research, and wrote the manuscript; X.Z., G.P., and L.R. performed functional studies and analysis; A.L. contributed to statistical design of the study; C.L. and T.L. performed and supervised manufacturing and characterization of VSTs; D.L. and J.F. performed data collection and analysis; J.A.C., S.T., C.D., J.W., Y.M.W., C.M.B., P.J.H., and M.D.K. provided vital conceptual insights for study design, assisted with study subject accrual and data collection, and provided scientific support; and all authors reviewed and edited the manuscript.
Conflict-of-interest disclosure: C.M.B. is on the advisory board for Cellectis and is on the scientific advisory boards for Catamaran Bio and Mana Therapeutics with stock or ownership; is on the board of directors for Caballeta Bio with stock options; and has stock in Neximmune and Torque Therapeutics. P.J.H. is a cofounder and on the board of directors of Mana Therapeutics, on the scientific advisory board of Cellevolve, and has intellectual property related to virus-specific T cells. S.M.D. has a US pending patent application under review, received research support from Alexion Pharmaceuticals and consultancy from Novartis (unrelated to this work). All other authors declare no competing financial interests.
Correspondence: Jeremy D. Rubinstein, Division of Oncology, Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Avenue, Cincinnati, OH 45229, MLC 7018; e-mail: Jeremy.rubinstein@cchmc.org.