

Key Points
Ectopic expression of RUNX1 induces binding site–directed DNA demethylation, in which hematopoietic gene promoters are included.
RUNX1 binding sites are enriched in demethylated regions during hematopoietic development.

Abstract
RUNX1 is an essential master transcription factor in hematopoietic development and plays important roles in immune functions. Although the gene regulatory mechanism of RUNX1 has been characterized extensively, the epigenetic role of RUNX1 remains unclear. Here, we demonstrate that RUNX1 contributes DNA demethylation in a binding site–directed manner in human hematopoietic cells. Overexpression analysis of RUNX1 showed the RUNX1-binding site–directed DNA demethylation. The RUNX1-mediated DNA demethylation was also observed in DNA replication–arrested cells, suggesting an involvement of active demethylation mechanism. Coimmunoprecipitation in hematopoietic cells showed physical interactions between RUNX1 and DNA demethylation machinery enzymes TET2, TET3, TDG, and GADD45. Further chromatin immunoprecipitation sequencing revealed colocalization of RUNX1 and TET2 in the same genomic regions, indicating recruitment of DNA demethylation machinery by RUNX1. Finally, methylome analysis revealed significant overrepresentation of RUNX1-binding sites at demethylated regions during hematopoietic development. Collectively, the present data provide evidence that RUNX1 contributes site specificity of DNA demethylation by recruitment of TET and other demethylation-related enzymes to its binding sites in hematopoietic cells.
Introduction
RUNX1 is an essential master transcription factor (TF) during hematopoietic development. All blood cell types differentiate from pluripotent stem cells, such as embryonic stem cells and induced pluripotent stem cells (iPSCs), through hematopoietic stem cells in vitro and in vivo.1 RUNX1-deficient mouse embryonic stem cells do not commit to the hematopoietic lineage.2 In addition, RUNX1 mutations and chromosomal truncations are frequently observed in acute myeloid leukemia.3,4 Thus, RUNX1 plays a pivotal role in normal hematopoietic development.
RUNX1 was identified as a transcriptional activator that regulates expression of downstream hematopoietic genes, such as interleukin-3, granulocyte-macrophage colony-stimulating factor, and PU.1 (SPI1).5-7 In addition, RUNX1 interacts with either activation cofactors, such as p300 and CBP (Creb Binding Protein), histone acetyltransferases,8,9 or repression cofactors such as SUV39H1, a histone methyltransferase.10 RUNX1 has also been shown to enhance EBF-derived DNA demethylation at the mb-1 promoter region.11 Accordingly, RUNX1 appears to contribute to not only direct gene activation but also epigenetic regulation by interacting with functional epigenetic enzymes.
DNA methylation is a crucial epigenetic modification because methylation of cytosine residues in cytosine guanine dinucleotide (CpG) dinucleotides of gene promoters represses transcription by prevention of TF binding, and therefore, DNA demethylation is fundamental for cell type–specific gene expression.12 DNA methylation is precisely regulated during cell differentiations, and abnormal DNA methylation is frequently observed in cancers. Hence, DNA methylation inhibitors such as 5-aza-2′-deoxycytidine are expected to be anticancer drugs.
De novo methylation of unmethylated cytosines is performed by DNA methyltransferase (DNMT) 3a/b, and the methylated status is inherited to daughter cells by DNMT1.13-15 In contrast, DNA demethylation occurs through passive16 and active17 mechanisms. Specifically, passive demethylation occurs after dilution of hemimethylated cytosines during DNA replication in the absence of DNMT1 activity, whereas multiple enzymes perform active DNA demethylation in a DNA replication-independent manner. Among these mechanisms, ten-eleven translocation methylcytosine dioxygenase (TET) oxidizes methylated cytosine nucleotides to hydroxyethyl cytosine, formyl cytosine, and then carboxyl cytosine.18-20 Subsequently, formyl and carboxyl cytosines are recognized as abnormal bases that are replaced by following base excision repair (BER) by thymine–DNA glycosylase (TDG), GADD45A, and various other proteins.21
During hematopoietic development, spatiotemporal changes in DNA methylation occur at the genome-wide level.22 Recently, several TFs, which play important roles in cellular differentiations, were reported to induce DNA demethylation by interacting with TET proteins.23-26 However, it has not been elucidated whether RUNX1 also contributes to regulating DNA methylation. Here, we demonstrate that RUNX1 contributes site-directed DNA demethylation by recruitment of DNA demethylation enzymes. Our findings provide evidence of a novel epigenetic function of RUNX1 and its potential contribution to hematopoietic development.
Methods
Cells and cell culture
Experiments were performed with human iPSCs (RIKEN BRC, HPS0002), CD34+ hematopoietic cells (Lonza, Basel, Switzerland; 2M-101c), CD14+ monocytes (MON; Lonza, 2W-400C), CD3+ T cells (HemaCare BioResearch, Van Nuys, CA; PB03C-2), CD19+ B cells (HemaCare BioResearch; PB19C-2), and peripheral blood mononuclear cells (Lonza, CC-2702). 293T cells were acquired from the BRC and cultured in Dulbecco’s modified Eagle medium (Wako, Osaka, Japan) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin. Jurkat cells were purchased from the BRC and cultured in RPMI-1640 (Wako) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin.
Methylation array analysis
Genomic DNA was isolated using a NucleoSpin Tissue Kit (Macherey-Nagel, Düren, Germany) and examined using an Infinium Humanmethylation450 BeadChip (Illumina, San Diego, CA) according to the manufacturer’s instructions. Data were quantile-normalized, and M values were calculated using the lumi Bioconductor package. CpGs that were differentially methylated between samples (DMCs) were selected using a cutoff M-value difference (ΔM) of >2.
Overrepresentation analyses of TF-binding motif
The TFBMs were searched in ±5-kbp regions from DMCs using the Bioconductor Biostrings package based on the hg19 genomic sequence. Motifs were counted in a 100-bp window sliding by 50-bp steps in ±200-bp regions from DMCs. Comparisons with the same numbers of randomly selected probes were performed using an exact test for the Poisson distribution model (P < .00001). Enrichment scores were then computed for significantly differing motifs using the following formula:
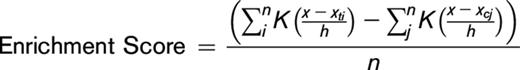
Here, i and j represent identified motifs in DMC regions and randomly selected controls, respectively, and xt and xc indicate motif positions in DMC regions and randomly selected controls, respectively. The smoothing parameter h was set at 50, and K was calculated as a Gaussian kernel function, as follows:

The significance of the enrichment is further assessed by positional profile: whether the peak of enrichment score is greater than barrier of Q3 + interquartile range × 3 in ±5-kbp regions.
ChIP-sequencing
Chromatin immunoprecipitation (ChIP) assays were performed as described previously27 using antibodies shown in supplemental Table 5. ChIPed DNA was subjected to library construction using a Library Preparation Kit (KAPA Biosystems, Wilmington, MA) and sequenced using the HiSequation 2500 system (Illumina). FASTQ data were mapped to human hg19 using Bowtie2. Peak calling and visualization were performed using MACS228 (cutoff P = .0001) and Ngsplot.
Bisulfite sequencing
Bisulfite conversion was performed using an EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA), and the target genomic regions were amplified using Epi-Taq HS (Takara Bio, Shiga, Japan) with the primers shown in supplemental Table 6. Polymerase chain reaction (PCR) products were cloned into pTA2 plasmids (Toyobo, Osaka, Japan), and 24 clones per target were sequenced. Visualization and statistical analysis were performed using QUMA.
Lentivirus preparation and transduction
The lentiviral vector CSII-EF-RfA was kindly provided by H. Miyoshi (RIKEN BRC), and puromycin-resistance gene fragment was inserted into the CSII-EF-RfA (CSII-EF-RfA-IRES2-Puro). Entry clone of RUNX1 was synthesized by Codon Devices and of TET2 and TET3 were acquired from the ORFeome collection (HsCD00377074) and GeneCopoeia (GC-H2292), respectively. The RUNX1 gene was subcloned into CSII-EF-RfA-Bsd,29 and TET2 and TET3 genes were subcloned into CSII-EF-RfA-Puro. Lentiviral vector preparation and transduction were then performed as described previously.29 Following transfection, antibiotic selection was performed using 2 μg/mL puromycin or 20 μg/mL blasticidin for 1 week.
qRT-PCR
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analyses were performed as described previously29 with the primers shown in supplemental Table 6. Gene expression was evaluated by ΔΔCt method compared with mock control sample.
Mitomycin C treatment
Confluent 293T cells were treated with mitomycin C at a final concentration of 5 μg/mL for 3 hours at 37°C, washed in phosphate-buffered saline twice, and then transduced with the RUNX1 lentiviral vector and cultured for 7 days.
3-Aminobenzamide treatment
Cells were plated at 1 × 104cells per well to 6-well plates. One day after the cell plating, the medium was replaced with fresh medium supplemented with 10 mM 3-aminobenzamide and RUNX1 lentivirus vector or mock control. After the medium replacement, the cells were cultured for 7 days.
qMSP
DNA was extracted using the NucleoSpin Tissue Kit, and the DNA methylation status was analyzed by quantitative methylation-specific PCR (qMSP) as described previously,30 with the primers shown in supplemental Table 6.
Co-IP and western blot
Cells were lysed in lysis buffer (50 mM Tris-HCl [pH 7.4], 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, Protease Inhibitor Cocktail). Antibodies were incubated with protein G Dynabeads (Thermo Fisher Scientific, Waltham, MA) for 1 hour, incubated with cell lysates at 4°C overnight, and washed twice in Tris-buffered saline washing buffer (0.05% IGEPA CA-630). Bead-bound proteins were eluted in elution buffer. Western blot was performed as described previously31 using the primary antibodies listed in supplemental Table 5 and a VeriBlot (Abcam, Cambridge, UK) IP secondary antibody.
HT immunoprecipitation analyses
HaloTag-fused expression vectors pFC14K-RUNX1, pFC14K-TDG, pFN21A-TET2, pFN21A-TET3, and pFN21A-GADD45A, expression vectors pCDNA3.1/V5-RUNX1, pCDNA3.1/V5-TDG, and pCDNA3.1/V5-GADD45A, and the V5-tag fused expression vector pCDNA3.2/nV5-TET2 and pCDNA3.2/nV5-TET3 were transfected into 293T cells using Fugne HD (Promega, Madison, WI) according to the manufacturer’s instructions. Cells were collected at 48 hours after transfection and high-throughput (HT)-based coimmunoprecipitation (co-IP) assays were performed using the HaloTag Mammalian Pull-Down System (Promega) according to the manufacturer’s instructions, followed by western blot.
Construction of deletion mutants
Entry clones of RUNX1 deletion mutants were constructed from a full-length RUNX1 by inverse PCR with the primers shown in supplemental Table 6 and were then subcloned into pCDNA3.2/nV5-DEST (Thermo Fisher Scientific).
RUNX1 knockdown
pLKO.1 shRUNX1 puro (#45816) and empty pLKO.1 puro (#8453) plasmids were purchased from Addgene. Lentiviral vector preparation and transduction were performed as described above. Knockdown efficiency was confirmed by qRT-PCR (supplemental Table 6).
Results
Ectopic expression of RUNX1 induces genome-wide binding site-directed DNA demethylation
To investigate whether RUNX1 is involved in DNA methylation regulation, we performed overexpression analyses in 293T cells that do not express RUNX1. Following ectopic expression of RUNX1 or the mock control by lentiviral vectors in 293T cells (293T-RUNX1 and 293T-mock, respectively), single-base resolution DNA methylation arrays were used to characterize global DNA methylation changes between 293T-RUNX1 and 293T-mock cells (Figure 1A). Comparing M-values, a statistical metric for log-scale methylation levels32 of 293T-RUNX1 cells with that of 293T-mock cells, we identified 145 and 59 demethylated and methylated CpGs (Figure 1B; supplemental Table 1). Interestingly, the RUNX1 promoter region was demethylated by the RUNX1 overexpression, suggesting an autofeedback loop, which consists of transcription and epigenetic alterations.

Induction of DNA demethylation by RUNX1 overexpression. (A) Confirmation of RUNX1 overexpression by western blotting. (B) Scatter plot showing DMCs caused by RUNX1 overexpression in 293T cells. x- and y-axes show M-values for 293T-mock and RUNX1-overexpressing 293T (293T-RUNX1) cells, respectively. Dashed lines represent ΔM borders of >2. Green, purple, and gray dots represent significantly methylated, demethylated, and insignificant probes, respectively. Numbers of DMCs are shown in the upper left (methylated) and bottom right (demethylated). Larger purple and gray dots represent targets and controls of qMSP analysis (Figures 2B and 4G; supplemental Figure 2). An enlarged view of the area surrounded by dashed red lined is shown in the left, and probe identifications of the targets and controls of qMSP analysis are labeled. (C) Distribution of enrichment scores for RUNX1-binding motifs within ±5000 bp of demethylated CpGs in RUNX1-overexpressing 293T cells. x- and y-axes indicate distance from probe CpG position and enrichment score, respectively. (D) ZENBU browser screenshots showing typical relationships between RUNX1 binding and the demethylated CpGs at cg07236781 (left) and cg03333149 (right) regions in RUNX1-overexpressing 293T cells. Demethylated CpG tracks show positions of demethylated CpG. ChIP-seq peaks and ChIP-seq tracks show the peak positions and tag per million (tpm), respectively. (E) A histogram showing distribution of RUNX1 ChIP-seq peaks in RUNX1-overexpressing 293T cells around demethylated CpGs (pink) and randomly selected probes (blue). Overlapped regions are shown as purple. x- and y-axes show distance from CpG (bp) and frequency of ChIP-seq peak, respectively. (F) DNA methylation patterns of PTPN22 and RUNX3 regions were determined using bisulfite sequencing in mock vector overexpressing 293T cells (293T-mock) and RUNX1-overexpressing 293T cells (293T-RUNX1). Horizontal lines show sequencing results for each subclone. Arrows represent positions of demethylated probes that were identified by methylation arrays. Circles represent cytosines of CpGs: black, methylated; white, unmethylated. Significant demethylation: *P < .05; **P < .01. Percentages and P values are shown at the bottom right. Percentages of methylated cytosines among all cytosines in the target region of all measured subclones were compared using Fisher’s exact test.
Induction of DNA demethylation by RUNX1 overexpression. (A) Confirmation of RUNX1 overexpression by western blotting. (B) Scatter plot showing DMCs caused by RUNX1 overexpression in 293T cells. x- and y-axes show M-values for 293T-mock and RUNX1-overexpressing 293T (293T-RUNX1) cells, respectively. Dashed lines represent ΔM borders of >2. Green, purple, and gray dots represent significantly methylated, demethylated, and insignificant probes, respectively. Numbers of DMCs are shown in the upper left (methylated) and bottom right (demethylated). Larger purple and gray dots represent targets and controls of qMSP analysis (Figures 2B and 4G; supplemental Figure 2). An enlarged view of the area surrounded by dashed red lined is shown in the left, and probe identifications of the targets and controls of qMSP analysis are labeled. (C) Distribution of enrichment scores for RUNX1-binding motifs within ±5000 bp of demethylated CpGs in RUNX1-overexpressing 293T cells. x- and y-axes indicate distance from probe CpG position and enrichment score, respectively. (D) ZENBU browser screenshots showing typical relationships between RUNX1 binding and the demethylated CpGs at cg07236781 (left) and cg03333149 (right) regions in RUNX1-overexpressing 293T cells. Demethylated CpG tracks show positions of demethylated CpG. ChIP-seq peaks and ChIP-seq tracks show the peak positions and tag per million (tpm), respectively. (E) A histogram showing distribution of RUNX1 ChIP-seq peaks in RUNX1-overexpressing 293T cells around demethylated CpGs (pink) and randomly selected probes (blue). Overlapped regions are shown as purple. x- and y-axes show distance from CpG (bp) and frequency of ChIP-seq peak, respectively. (F) DNA methylation patterns of PTPN22 and RUNX3 regions were determined using bisulfite sequencing in mock vector overexpressing 293T cells (293T-mock) and RUNX1-overexpressing 293T cells (293T-RUNX1). Horizontal lines show sequencing results for each subclone. Arrows represent positions of demethylated probes that were identified by methylation arrays. Circles represent cytosines of CpGs: black, methylated; white, unmethylated. Significant demethylation: *P < .05; **P < .01. Percentages and P values are shown at the bottom right. Percentages of methylated cytosines among all cytosines in the target region of all measured subclones were compared using Fisher’s exact test.
RUNX1 binds to a specific consensus motif with a core sequence motif [TGTGG(TTT/TCA)].33 Therefore, we hypothesized that, if RUNX1 is involved in regulation of DNA methylation in a binding site-directed manner, the RUNX1-binding motif will be overrepresented at differentially methylated regions in RUNX1-overexpressing cells. To test this hypothesis, we performed TF-binding motif (TFBM) overrepresentation analyses by using an algorithm that was developed in our laboratory (supplemental Figure 1A, see “Methods”) in RUNX1-overexpressing cells. The result revealed significant overrepresentation of the RUNX1-binding motif in the immediate vicinity of the demethylated regions (P = 1 × 10−30, exact test for Poisson distribution; Figure 1C) but not in that of the methylated regions (supplemental Figure 1B). This RUNX1-binding motif overrepresentation at demethylated regions by RUNX1 overexpression was validated in an independent analysis by using a different methylation array platform (Human MethylationEpic BeadChip [Illumina]) (P < 1 × 10−100, exact test for Poisson distribution) (supplemental Figure 1C-D).
To further confirm link between DNA demethylation and RUNX1 binding, we also performed a chromatin immunoprecipitation-sequencing (ChIP-seq) analysis of RUNX1 in RUNX1-overexpressing 293T cells. The ChIP-seq analysis identified 96 158 peaks. Of these, 391 and 265 peaks were harbored in ±5 kb from demethylated and randomly selected probes, respectively, showing significant overrepresentation of RUNX1 ChIP peaks around the demethylated CpGs (P = 1.93 × 10−13, exact test for Poisson distribution) (Figure 1D-E).
In subsequent bisulfite sequencing analyses of 2 demethylated regions, cg00041401 (PTPN22) and cg07236781 (RUNX3), in RUNX1-overexpressing cells, both demethylated CpGs and neighboring CpGs were demethylated (Figure 1F).
Overall, these results indicate that RUNX1 induces DNA demethylation at its binding regions.
RUNX1-mediated DNA demethylation occurs via a combination of DNA replication-dependent and -independent mechanisms
The 2 mechanisms of DNA demethylation (active and passive) are DNA replication-independent and -dependent, respectively. To determine which mechanism is responsible for RUNX1-mediated DNA demethylation, we examined RUNX1-mediated DNA demethylation in DNA replication-arrested 293T cells using qMSP. Proliferation of 293T cells was arrested by treatment with 5 μg/mL mitomycin C (Figure 2A), followed by RUNX1 overexpression and qMSP analyses of 3 RUNX1-mediated DNA demethylation target sites that were randomly selected from the above methylation array results (Figure 1B). qMSP revealed that, although the demethylation was weaker than in mitomycin C untreated 293T cells, 2 RUNX1 targets were significantly demethylated and 1 was slightly demethylated, albeit not statistically significant, by RUNX1 overexpression compared with mock control overexpression in DNA replication-arrested 293T cells (Figure 2B). Conversely, 2 randomly selected negative control regions were not demethylated (Figure 2B). Furthermore, treatment of an inhibitor of active DNA demethylation, 3-aminobenzamide,34 significantly reduced the RUNX1-mediated DNA demethylation (supplemental Figure 2). These results suggest that both active and passive mechanisms contribute to RUNX1-mediated DNA demethylation.

RUNX1-mediated DNA demethylation in mitomycin C–treated 293T cells. (A) 293T cell growth after mitomycin C (MMC) treatment. 293T cells were treated with the indicated concentrations of MMC, and then cell densities were measured after 1, 3, and 6 days. Data are presented as means ± standard deviation (SD) of 3 biological replicates. (B) qMSP analysis of 3 RUNX1-mediated DNA demethylation target regions (RUNX1 targets) and 2 randomly selected negative control regions (Random) in mock vector–overexpressing, RUNX1-overexpressing, MMC-treated mock vector–overexpressing, and MMC-treated RUNX1-overexpressing 293T cells, respectively (Mock, RUNX1, MMC-mock, and MMC-RUNX1). The vertical axis represents ΔCt (unmethylated-specific primer-methylated-specific primer). Error bars represent SD. Asterisks denote significant difference: *P < .05, **P < .01; N.S., not significant. The experiments were performed in 3 biological replicates.
RUNX1-mediated DNA demethylation in mitomycin C–treated 293T cells. (A) 293T cell growth after mitomycin C (MMC) treatment. 293T cells were treated with the indicated concentrations of MMC, and then cell densities were measured after 1, 3, and 6 days. Data are presented as means ± standard deviation (SD) of 3 biological replicates. (B) qMSP analysis of 3 RUNX1-mediated DNA demethylation target regions (RUNX1 targets) and 2 randomly selected negative control regions (Random) in mock vector–overexpressing, RUNX1-overexpressing, MMC-treated mock vector–overexpressing, and MMC-treated RUNX1-overexpressing 293T cells, respectively (Mock, RUNX1, MMC-mock, and MMC-RUNX1). The vertical axis represents ΔCt (unmethylated-specific primer-methylated-specific primer). Error bars represent SD. Asterisks denote significant difference: *P < .05, **P < .01; N.S., not significant. The experiments were performed in 3 biological replicates.
TET enhances RUNX1-mediated DNA demethylation
Although TET proteins (TET1, TET2, and TET3) play key roles in active DNA demethylation,20 expressions of these genes are low to middle in HEK293 cells (supplemental Figure 3A-B). Therefore, we hypothesized that overexpression of TETs enhance the RUNX1-mediated DNA demethylation. DNA methylation array analyses were performed in TET2- or TET3- and RUNX1-co-overexpressing cells (293T-T2R, 293T-T3R, respectively) (Figure 3A-B), because TET2 and TET3 are expressed in hematopoietic lineage cells as well as RUNX1. Because TET2 or TET3 single overexpression induced DNA methylation alterations in the absence of RUNX1, which can be considered nonspecific effects (supplemental Figure 3C-D), we compared 293T-T2R and 293T-T3R with TET2- and TET3-overexpressing 293T cells (293T-T2 and 293T-T3, respectively) to clarify the RUNX1 overexpression effect. These comparative analyses showed that RUNX1 overexpression together with TET2 or TET3 expression led to more demethylated CpGs (565 and 357, respectively) compared with RUNX1 single overexpression (Figure 3C). Of these demethylated CpGs, 97 (66.9%, P = 2.2 × 10−16; χ2 test) and 83 (57.2%, P = 2.2 × 10−16; χ2 test) in 293T-T2R and 293T-T3R cells overlapped with those in RUNX1-overexpressing cells, respectively, and 251 (44.4% and 70.3%) demethylated CpGs in 293T-T2R and 293T-T3R cells overlapped each other. In the proximal regions of these demethylated CpGs, RUNX1-binding motifs were significantly overrepresented (TET2 vs TET2 + RUNX1; P = 1.23 × 10−161, TET3 vs TET3 + RUNX1; P < 1 × 10−500), ruling out the possibility of nonspecific demethylation (Figure 3D). Taken together, these data indicate that TET2 and TET3 enhance RUNX1-mediated DNA demethylation.

TETs enhance RUNX1-mediated DNA demethylation. (A) Confirmation of RUNX1, TET2, and TET3 overexpression in mock vector–overexpressing (293T-mock), RUNX1-overexpressing (293T-RUNX1), TET2-overexpressing (293T-TET2), TET3-overexpressing (293T-TET3), TET2 and RUNX1–co-overexpressing (293T-TET2-RUNX1), and TET3 and RUNX1–co-overexpressing (293T-TET3-RUNX1) 293T cells by western blotting. Immunoblotting for GAPDH is internal control. (B) Confirmation of RUNX1 (left), TET2 (middle), and TET3 (right) overexpression by qRT-PCR. y-axis represents average log2 fold change (FC) compared with mock vector–transduced cells. The error bars were SD. The experiments were performed in 3 biological replicates. (C) Overlap of demethylated CpGs between TET2-overexpressing and TET2 + RUNX1–overexpressing cells (TET2 vs TET2 + RUNX1) (blue), between TET3-overexpressing and TET3 + RUNX1–overexpressing cells (TET3 vs TET3 + RUNX1) (green), and between mock- and RUNX1-overexpressing cells (mock vs RUNX1) (red). The size of each circle represents total number of the demethylated CpGs. (D) Distribution of enrichment scores for RUNX1-binding motif within ±5000 bp from demethylated CpGs in TET2- and RUNX1-overexpressing 293T cells (red line), TET3- and RUNX1-overexpressing 293T cells (blue line), and RUNX1-overexpressing 293T cells (gray line). x- and y-axes show distance from CpG (bp) and enrichment score of RUNX1-binding motif, respectively.
TETs enhance RUNX1-mediated DNA demethylation. (A) Confirmation of RUNX1, TET2, and TET3 overexpression in mock vector–overexpressing (293T-mock), RUNX1-overexpressing (293T-RUNX1), TET2-overexpressing (293T-TET2), TET3-overexpressing (293T-TET3), TET2 and RUNX1–co-overexpressing (293T-TET2-RUNX1), and TET3 and RUNX1–co-overexpressing (293T-TET3-RUNX1) 293T cells by western blotting. Immunoblotting for GAPDH is internal control. (B) Confirmation of RUNX1 (left), TET2 (middle), and TET3 (right) overexpression by qRT-PCR. y-axis represents average log2 fold change (FC) compared with mock vector–transduced cells. The error bars were SD. The experiments were performed in 3 biological replicates. (C) Overlap of demethylated CpGs between TET2-overexpressing and TET2 + RUNX1–overexpressing cells (TET2 vs TET2 + RUNX1) (blue), between TET3-overexpressing and TET3 + RUNX1–overexpressing cells (TET3 vs TET3 + RUNX1) (green), and between mock- and RUNX1-overexpressing cells (mock vs RUNX1) (red). The size of each circle represents total number of the demethylated CpGs. (D) Distribution of enrichment scores for RUNX1-binding motif within ±5000 bp from demethylated CpGs in TET2- and RUNX1-overexpressing 293T cells (red line), TET3- and RUNX1-overexpressing 293T cells (blue line), and RUNX1-overexpressing 293T cells (gray line). x- and y-axes show distance from CpG (bp) and enrichment score of RUNX1-binding motif, respectively.
RUNX1 physically interacts with TET, TDG, and GADD45A
We next examined physical interactions between RUNX1 and TET2. We performed co-IP assays followed by western blot analyses in the Jurkat T-lymphocyte line. Jurkat expressed RUNX1 and TET2 (supplemental Figure 4A), and the RUNX1-binding motif was overrepresented in demethylated regions compared with iPSCs (P < 1 × 10−500, exact test for Poisson distribution) (supplemental Figure 4B). Therefore, we used Jurkat as a model hematopoietic cell line for molecular analyses. Co-IP followed by western blot showed that RUNX1 and TET2 proteins coprecipitated each other (Figure 4A), indicating an endogenous physical interaction between RUNX1 and TET2. HaloTag (HT)-based co-IP assays also confirmed the interaction between RUNX1 and TET2 in 293T cells (Figure 4B; supplemental Figure 4C). The HT-based co-IP assay further revealed interactions between RUNX1 and BER pathway enzymes TDG and GADD45A35 that are involved in active DNA demethylation (Figure 4C-D; supplemental Figure 4D-E). We also detected interaction between RUNX1 and TET3 by using the HT-based co-IP assay in 293T cells (supplemental Figure 4F-G). Cumulatively, these results suggest that RUNX1 forms a DNA demethylation machinery complex by interacting with enzymes involved in active DNA demethylation.

Physical interactions between RUNX1 and DNA demethylation proteins. (A) Co-IP of endogenous RUNX1 or TET2 followed by western blotting in Jurkat. Input indicates 0.05%, 0.1%, and 1% (from left) of nonimmunoprecipitated cell lysate; IgG, control IP with isotype antibody; IB, immunoblot. (B) HT-based co-IP of HaloTag-fused TET2 (TET2-HT) in TET2-HT and RUNX1 co-overexpressing 293T cells (left) and of HaloTag (HT-ctrl) in HT-ctrl and RUNX1 co-overexpressing 293T cells (right) followed by western blot for RUNX1 protein. (C) HT-based co-IP of HaloTag-fused RUNX1 (RUNX1-HT) in RUNX1-HT and TDG co-overexpressing 293T cells (left), of TET2-HT in TET2-HT and TDG co-overexpressing 293T cells (center), and of HT-ctrl in HT-ctrl and TDG co-overexpressing 293T cells (right) followed by western blotting for TDG protein. (D) HT-based co-IP of RUNX1-HT in RUNX1-HT and GADD45A co-overexpressing 293T cells and of HT-ctrl in HT-ctrl and GADD45A co-overexpressing 293T cells followed by western blotting for GADD45A protein. (E) Schematic representation of RUNX1 deletion mutants. RUNT (red) and TA (light red) denote RUNT DNA-binding and transactivation (TA) domains, respectively. The deleted portions are shown as ranges of amino acid position in the left of each schematic. (F) HT-based co-IP of TET2-HT in TET2-HT and the RUNX1 deletion mutant co-overexpressing 293T cells followed by western blot for V5-tag. The y-axis represents molecular weight. (G) qMSP analysis in RUNX1 deletion mutants overexpressing 293T cells for 3 RUNX1-mediated DNA demethylation target regions (blue) and 2 randomly selected control regions (light blue). Horizontal axes represent methylation percentages with SD. The experiments were performed in 3 biological replicates. WT, wild type.
Physical interactions between RUNX1 and DNA demethylation proteins. (A) Co-IP of endogenous RUNX1 or TET2 followed by western blotting in Jurkat. Input indicates 0.05%, 0.1%, and 1% (from left) of nonimmunoprecipitated cell lysate; IgG, control IP with isotype antibody; IB, immunoblot. (B) HT-based co-IP of HaloTag-fused TET2 (TET2-HT) in TET2-HT and RUNX1 co-overexpressing 293T cells (left) and of HaloTag (HT-ctrl) in HT-ctrl and RUNX1 co-overexpressing 293T cells (right) followed by western blot for RUNX1 protein. (C) HT-based co-IP of HaloTag-fused RUNX1 (RUNX1-HT) in RUNX1-HT and TDG co-overexpressing 293T cells (left), of TET2-HT in TET2-HT and TDG co-overexpressing 293T cells (center), and of HT-ctrl in HT-ctrl and TDG co-overexpressing 293T cells (right) followed by western blotting for TDG protein. (D) HT-based co-IP of RUNX1-HT in RUNX1-HT and GADD45A co-overexpressing 293T cells and of HT-ctrl in HT-ctrl and GADD45A co-overexpressing 293T cells followed by western blotting for GADD45A protein. (E) Schematic representation of RUNX1 deletion mutants. RUNT (red) and TA (light red) denote RUNT DNA-binding and transactivation (TA) domains, respectively. The deleted portions are shown as ranges of amino acid position in the left of each schematic. (F) HT-based co-IP of TET2-HT in TET2-HT and the RUNX1 deletion mutant co-overexpressing 293T cells followed by western blot for V5-tag. The y-axis represents molecular weight. (G) qMSP analysis in RUNX1 deletion mutants overexpressing 293T cells for 3 RUNX1-mediated DNA demethylation target regions (blue) and 2 randomly selected control regions (light blue). Horizontal axes represent methylation percentages with SD. The experiments were performed in 3 biological replicates. WT, wild type.
To identify the TET2-binding domain of RUNX1, we performed HT-based co-IP analyses using RUNX1 deletion mutants. Because the DNA-binding domain of RUNX1 (RUNT domain) is necessary for sequence-specific binding to DNA, we designed 4 deletion mutants with intact RUNT domains (Figure 4E). V5-tagged RUNX1 deletion mutants and HT-fused TET2 were overexpressed in 293T cells followed by HT co-IP analyses. Three of the 4 RUNX1 mutants clearly coprecipitated with TET2, whereas the Δ300-480 RUNX1 mutant showed a very weak interaction (Figure 4F), suggesting that RUNX1 binds to TET via a region from 300 to 480 aa. To confirm disruption of the RUNX1-mediated DNA demethylation activity in the Δ300-480 RUNX1 mutant, we performed qMSP analysis. The results showed a lack of demethylation activity in the Δ300-480 RUNX1 mutant (Figure 4G), supporting the interaction of RUNX1 with TET2 through the 300-480 aa region. Overall, these results indicate that the interaction with TET2 is necessary for RUNX1-mediated DNA demethylation.
Genome-wide colocalization of RUNX1 and TET2
To determine whether RUNX1 and TET2 colocalize in the same chromatin regions, we performed ChIP-seq analyses of RUNX1 and TET2 in Jurkat. A total of 25 881 625 and 9 466 629 reads were obtained from RUNX1 and TET2 ChIP-seqs, respectively, which were mapped onto the hg19 human genome (Figure 5A). We identified 50 334 and 126 184 peaks in RUNX1 and TET2 ChIP-seqs, respectively. Of these peaks, 40 982 of RUNX1 peaks (81.4%) and 38 345 of TET2 peaks (30.4%) overlapped each other, indicating that a substantial number of RUNX1-binding sites overlap with TET2-binding sites (Figure 5B). Genome-wide analysis also showed enrichment of TET2 reads around the RUNX1 ChIP peaks, which was significantly reduced by RUNX1 knockdown (P = .028, Student t test) (Figure 5C-E; supplemental Figure 5A). The enrichment of TET2 reads around RUNX1 ChIP peaks was validated in human peripheral blood mononuclear cells (supplemental Figure 5B). These results suggest that TET2 colocalizes with RUNX1 in a RUNX1-dependent manner. Furthermore, heatmap analysis revealed that both RUNX1 and TET2 ChIP-seq reads were highly enriched at transcription start sites (TSSs) (Figure 5F) and CpG islands (Figure 5G) as described previously.36,37 Collectively, these results suggest RUNX1:TET2 colocalization at gene regulatory regions such as promoters and CpG islands.
![Figure 5. ChIP-seq analyses of RUNX1 and TET2. (A) A ZENBU browser screenshot showing a typical relationship between RUNX1 and TET2 ChIP-seq data, and the methylation level. The DNA methylation track is the M-value measured by the methylation array. Green and purple bars represent positive and negative M-values, respectively. ChIP-seq tracks show the tpm. (B) Overlap between RUNX1 (blue) and TET2 (red) ChIP-seq peaks. (C) Distribution of TET2 ChIP-seq reads around RUNX1 ChIP-seq peaks. The x- and y-axes indicate ±3-kbp genomic region from RUNX1 ChIP-seq peaks and read count per million mapped read, respectively. The solid line and shaded area render average and standard error of read count per million mapped reads. The experiment was done in 2 biological replicates. (D) Distribution of TET2 ChIP-seq reads around RUNX1 ChIP-seq peaks in RUNX1-knockdown Jurkat (RUNX1_shRNA [short hairpin RNA], green line) and negative control (NC_shRNA, red line). The x- and y-axes indicate ±3-kbp genomic region from the summit of RUNX1 ChIP-seq peaks and read count per million mapped read, respectively. The solid lines and shaded area render average and SD. The experiment was done in 2 biological replicates. (E) Total read count per million mapped read at ±3000-bp regions from RUNX1 ChIP-seq peaks. Red and green bars represent NC shRNA transduced Jurkat cells (NC_shRNA) and RUNX1-knockdown Jurkat cells (RUNX1_shRNA), respectively. Error bars represent SD. The asterisk denotes P < .05. The experiment was done in 2 biological replicates. (F-G) Distribution of RUNX1 and TET2 ChIP-seq reads around TSSs (±3 kb) (F) and CpG islands (CGI; ±3 kb) (G). The color key represents read counts per million. (H) Scatter plots showing relation between M-value and ChIP-seq peak heights of RUNX1 (top), TET2 (middle), and RUNX1 at regions with both RUNX1 and TET2 (bottom, RUNX1 at intersection). x- and y-axes represent M-value and −log10q value, respectively. The scatterplots are represented as blue smoothed shade density.](/view-large/figure/4448870/advances005710f5.jpeg)
ChIP-seq analyses of RUNX1 and TET2. (A) A ZENBU browser screenshot showing a typical relationship between RUNX1 and TET2 ChIP-seq data, and the methylation level. The DNA methylation track is the M-value measured by the methylation array. Green and purple bars represent positive and negative M-values, respectively. ChIP-seq tracks show the tpm. (B) Overlap between RUNX1 (blue) and TET2 (red) ChIP-seq peaks. (C) Distribution of TET2 ChIP-seq reads around RUNX1 ChIP-seq peaks. The x- and y-axes indicate ±3-kbp genomic region from RUNX1 ChIP-seq peaks and read count per million mapped read, respectively. The solid line and shaded area render average and standard error of read count per million mapped reads. The experiment was done in 2 biological replicates. (D) Distribution of TET2 ChIP-seq reads around RUNX1 ChIP-seq peaks in RUNX1-knockdown Jurkat (RUNX1_shRNA [short hairpin RNA], green line) and negative control (NC_shRNA, red line). The x- and y-axes indicate ±3-kbp genomic region from the summit of RUNX1 ChIP-seq peaks and read count per million mapped read, respectively. The solid lines and shaded area render average and SD. The experiment was done in 2 biological replicates. (E) Total read count per million mapped read at ±3000-bp regions from RUNX1 ChIP-seq peaks. Red and green bars represent NC shRNA transduced Jurkat cells (NC_shRNA) and RUNX1-knockdown Jurkat cells (RUNX1_shRNA), respectively. Error bars represent SD. The asterisk denotes P < .05. The experiment was done in 2 biological replicates. (F-G) Distribution of RUNX1 and TET2 ChIP-seq reads around TSSs (±3 kb) (F) and CpG islands (CGI; ±3 kb) (G). The color key represents read counts per million. (H) Scatter plots showing relation between M-value and ChIP-seq peak heights of RUNX1 (top), TET2 (middle), and RUNX1 at regions with both RUNX1 and TET2 (bottom, RUNX1 at intersection). x- and y-axes represent M-value and −log10q value, respectively. The scatterplots are represented as blue smoothed shade density.
ChIP-seq analyses of RUNX1 and TET2. (A) A ZENBU browser screenshot showing a typical relationship between RUNX1 and TET2 ChIP-seq data, and the methylation level. The DNA methylation track is the M-value measured by the methylation array. Green and purple bars represent positive and negative M-values, respectively. ChIP-seq tracks show the tpm. (B) Overlap between RUNX1 (blue) and TET2 (red) ChIP-seq peaks. (C) Distribution of TET2 ChIP-seq reads around RUNX1 ChIP-seq peaks. The x- and y-axes indicate ±3-kbp genomic region from RUNX1 ChIP-seq peaks and read count per million mapped read, respectively. The solid line and shaded area render average and standard error of read count per million mapped reads. The experiment was done in 2 biological replicates. (D) Distribution of TET2 ChIP-seq reads around RUNX1 ChIP-seq peaks in RUNX1-knockdown Jurkat (RUNX1_shRNA [short hairpin RNA], green line) and negative control (NC_shRNA, red line). The x- and y-axes indicate ±3-kbp genomic region from the summit of RUNX1 ChIP-seq peaks and read count per million mapped read, respectively. The solid lines and shaded area render average and SD. The experiment was done in 2 biological replicates. (E) Total read count per million mapped read at ±3000-bp regions from RUNX1 ChIP-seq peaks. Red and green bars represent NC shRNA transduced Jurkat cells (NC_shRNA) and RUNX1-knockdown Jurkat cells (RUNX1_shRNA), respectively. Error bars represent SD. The asterisk denotes P < .05. The experiment was done in 2 biological replicates. (F-G) Distribution of RUNX1 and TET2 ChIP-seq reads around TSSs (±3 kb) (F) and CpG islands (CGI; ±3 kb) (G). The color key represents read counts per million. (H) Scatter plots showing relation between M-value and ChIP-seq peak heights of RUNX1 (top), TET2 (middle), and RUNX1 at regions with both RUNX1 and TET2 (bottom, RUNX1 at intersection). x- and y-axes represent M-value and −log10q value, respectively. The scatterplots are represented as blue smoothed shade density.
To investigate differences in the DNA methylation levels between regions with RUNX1 and with TET2, we superimposed ChIP-seq data and methylation array data. RUNX1 tended to strongly bind to hypomethylated regions and weakly bind to hypermethylated regions (Figure 5H top). Interestingly, regions with TET2 tended not to harbor hypomethylation (Figure 5H middle). Therefore, regions with both RUNX1 and TET2 biased toward hypermethylation with weak RUNX1 binding (Figure 5H bottom). Taken together, the results suggest that colocalization of RUNX1 and TET2 is affected by DNA methylation level of their binding region.
RUNX1-binding sites are significantly overrepresented in demethylated regions during hematopoietic development
To investigate whether the RUNX1-mediated DNA demethylation is involved in hematopoietic development, we performed TFBM overrepresentation analysis of differentially methylated regions during hematopoietic development. DNA methylation arrays were used to characterize global DNA methylation changes during hematopoietic differentiation of iPSCs, CD34+ HPCs, CD14+ MONs, CD19+ B cells, and CD3+ TCs (Figure 6A). To identify differentially methylated CpGs, we compared M-values between iPSCs and HPCs (iPSCs-HPCs) and between HPCs and 3 terminally differentiated hematopoietic cell types (HPCs-MONs, HPCs-BCs, and HPCs-TCs). These analyses identified 24 289, 1814, 2360, and 11 733 methylated (Figure 6B green dots) and 32 434, 12 410, 10 278, and 26 376 demethylated (Figure 6B purple dots) CpGs in iPSC-HPCs, HPCs-MONs, HPCs-BCs, and HPCs-TCs, respectively (supplemental Table 2). Importantly, out of demethylated CpGs by the RUNX1 overexpression, 82 (56.6%) overlapped with demethylated CpGs in any of the hematopoietic differentiations (P = 1.58 × 10−106, χ2 test) (supplemental Table 1, “Overlap” column), including CpGs harbored in the promoter (from 1.5 kbp upstream of the TSS to the first exon) of hematopoietic genes, such as PTPN22, RUNX1, RUNX3, IL17RE, LSP1, and BST2 (Tetherin).38-44 Further characterization of the 82 overlapped demethylated regions by using Roadmap epigenetics datasets45 revealed that H3K4me1, an enhancer mark, was highly enriched at the demethylated regions in hematopoietic cell types, suggesting that RUNX1-mediated DNA demethylation predominantly targets enhancer regions (supplemental Figure 6A).

RUNX1-binding motif overrepresentation analysis of differentially methylated regions during hematopoietic development. (A) Differentiation hierarchy of hematopoietic development. (B) Scatter plots of M-values between induced pluripotent stem cells (iPSCs) and CD34+ hematopoietic progenitor cells (HPCs), between HPCs and CD14+ monocytes (MONs), between HPCs and CD19+ B cells (BC), and between HPCs and CD3+ T cells (TCs). Dashed black lines represent ΔM borders of >2. Green, purple, and gray dots represent significantly methylated, demethylated, and insignificant probes, respectively. Numbers of methylated and demethylated CpGs in each comparison are shown in the upper left and lower right of each plot, respectively. (C) Distribution of RUNX1-binding motif enrichment scores at ±5000 bp from DMCs. (D) Histogram showing distribution of RUNX1 ChIP-seq peaks in HPCs around demethylated regions of iPSC-HPCs (pink bars) and randomly sampled same number of probes (light blue bars). The overlapped regions are shown in purple.
RUNX1-binding motif overrepresentation analysis of differentially methylated regions during hematopoietic development. (A) Differentiation hierarchy of hematopoietic development. (B) Scatter plots of M-values between induced pluripotent stem cells (iPSCs) and CD34+ hematopoietic progenitor cells (HPCs), between HPCs and CD14+ monocytes (MONs), between HPCs and CD19+ B cells (BC), and between HPCs and CD3+ T cells (TCs). Dashed black lines represent ΔM borders of >2. Green, purple, and gray dots represent significantly methylated, demethylated, and insignificant probes, respectively. Numbers of methylated and demethylated CpGs in each comparison are shown in the upper left and lower right of each plot, respectively. (C) Distribution of RUNX1-binding motif enrichment scores at ±5000 bp from DMCs. (D) Histogram showing distribution of RUNX1 ChIP-seq peaks in HPCs around demethylated regions of iPSC-HPCs (pink bars) and randomly sampled same number of probes (light blue bars). The overlapped regions are shown in purple.
Subsequently, to assess TFBM overrepresentation in an unbiased way, we screened overrepresented TFBMs. Out of 113 human and mouse TFBMs, which are registered in the JASPAR CORE,46 29, 10, 7, and 12 were overrepresented at demethylated regions in iPSCs-HPCs, HPCs-MONs, HPCs-BCs, and HPCs-TCs, respectively, which include hematopoietic linage-specific TF responsible motifs, such as SPI1, RUNX1, SPIB, NR4A2, IRF1, IRF2, and AP-147-51 (supplemental Table 3). Notably, the RUNX1-binding motif was overrepresented at the demethylated regions of all differentiations (Figure 6C top). On the other hand, we identified 7 and 4 TFBMs at the methylated regions in iPSCs-HPCs, and HPCs-TCs, respectively, which did not include the RUNX1-binding motif (Figure 6C bottom; supplemental Table 4). The RUNX1-binding motif overrepresentation was positively correlated with RUNX1 expression, which is computed from the FANTOM5 database52 (R = 0.79) (supplemental Figure 6B). Furthermore, the RUNX1-binding motif overrepresentation in demethylated regions was reproduced in different donor samples of published methylation array data (GSE40790): between iPSCs (iGP91-02) and 2 lines of human peripheral blood CD34+ cells (CD34_1 and CD34_2), or human peripheral blood CD14+ cells (CD14) (supplemental Figure 6C). Thus, these results suggest that RUNX1 is 1 of the TFs that contribute to the DNA demethylation regulation in hematopoietic differentiation.
Because 1 TFBM sometimes corresponds to multiple TFs, we examined whether the RUNX1-binding motif overrepresentation at demethylated regions reflect actual RUNX1 binding. We compared the demethylated regions of iPSC-HPCs with RUNX1 ChIP-seq peaks of HPCs. We identified 8768 RUNX1-binding peaks in publically available RUNX1-ChIP-seq data of HPCs (GSE45144). Of these, 3130 peaks were located at ±5-kb regions from demethylated CpGs, which showed statistically significant overrepresentation compared with ±5-kb regions from randomly selected probes (P = 3.85 × 10−10, exact test for Poisson distribution), indicating the DNA demethylation links to RUNX1 binding (Figure 6D).
Collectively, these results suggest an involvement of RUNX1 in DNA demethylation at a subset of its binding site proximal regions at multiple hematopoietic development stages.
Discussion
In this study, we demonstrated that RUNX1 contributes site specificity of DNA demethylation by both active and passive mechanisms. In the active DNA methylation, we showed recruitment of DNA demethylation machinery by RUNX1. A possible mechanism of TF-mediated passive DNA demethylation is that DNMT1 recruitment is physically inhibited by TF binding,53,54 which may also occur in RUNX1-mediated DNA demethylation .In addition, hydroxymethyl-, formyl-, and carboxyl-cytosines, which are oxidized by RUNX1-mediated recruitment of TET, may block maintenance methylation during cell division, although it is still controversial.55-57
We revealed enhancement of RUNX1-mediated DNA demethylation by TET2 or TET3 co-overexpression. In the context of RUNX1 overexpression, RUNX1 proteins may be in excess compared with endogenous TET proteins because TET expression level is not high. Therefore, additional overexpression of TET may compensate for the imbalance of RUNX1 and TET proteins, resulting in enhancement of RUNX1-mediated DNA demethylation. Another possible mechanism of demethylation enhancement by TET overexpression may be increased accessibility of DNA. However, because we compared TET and RUNX1 co-overexpression with TET single overexpression to remove the effect of TET alone, this possibility can also be eliminated.
In zebrafish, Tet2 and Tet3 have overlapping functions in promotion of normal development.58 Furthermore, both TET2 and TET3 regulate GlcNAcylation by interacting with O-GlcNAc transferase.59 Our data indicate that both TET2 and TET3 enhance RUNX1-mediated DNA demethylation, and each effect is highly overlapped, supporting the functional overlap between TET2 and TET3.
Co-IP analyses showed interactions between RUNX1 and active DNA demethylation machinery enzymes. BER is a pivotal step in active DNA demethylation. Interestingly, RUNX1 regulates the expression of BER-related genes, including OGG1 and OLE.60,61 Thus, RUNX1-mediated DNA demethylation may be enhanced by a transcription-based auto-feedback loop.
ChIP-seq analyses revealed that TET2 was not associated with RUNX1 at hypomethylated genomic regions, although RUNX1 and TET2 were generally colocalized. This result suggests that TET2-RUNX1 complexes dissociate rapidly following to the completion of demethylation. A recent study showed that IDAX protein binds to both unmethylated CpG and TET2, and then downregulates TET2 protein expression, although the related mechanisms have not been described.62 This finding suggests the involvement of IDAX in the reduction of TET2 from demethylated regions.
The proposed mechanism of RUNX1-mediated demethylation is consistent with that of other reported TFs that induce DNA demethylation in a TET-dependent manner.23-26 Thus, TFs with a TET-dependent demethylation function may comprise a functional subclass. However, because a limited number of TFs have been identified in this subclass, it is unclear how major this subclass is. Therefore, whether this mechanism is a general function of TFs should be investigated further.
Our data showed that RUNX1-mediated DNA demethylation may be involved in hematopoietic development. Although it is widely established that RUNX1 is essential for hematopoietic commitment, and dysfunction of RUNX1 is linked to hematological diseases such as leukemia, the contribution of the demethylation function of RUNX1 to normal development and diseases is completely unknown. Therefore, physiological and pathological analyses of RUNX1-mediated DNA demethylation are of great interest.
DNA demethylation is an essential step for gene activation, changing “silenced state” to “poised state,”63 enabling additional TF binding.64 Thus, RUNX1-mediated DNA demethylation may unlock the silenced hematopoietic genes, and other factor(s) subsequently regulates the expression.
DNA methylation inhibitors such as 5-azacytidine are used to treat precancerous lesions in myelodysplastic syndrome, suggesting that aberrant DNA methylation is a major factor in oncogenesis. However, nonspecificity of DNA methylation inhibitors may lead to significant side effects. Because RUNX1 induces binding site-specific DNA demethylation, our findings may contribute to the development of highly specific epigenome drugs for cancer therapeutics that induce fewer adverse effects.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Dave Tang and Marina Lizio for helpful advice on bioinformatics analysis and the members of RIKEN Center for Life Science Technologies Division of Genomic Technologies.
This work was supported by a research grant from the Ministry of Education, Culture, Sport, Science, and Technology of Japan for the RIKEN Center for Life Science Technologies.
Authorship
Contribution: T.S., Y.H., and H.S. were instrumental in the conceptualization; T.S. oversaw the methodology and the software and wrote the original draft; T.S., Y.S., E.F., S.M., M.K., H.N., and S.E. oversaw the investigation; H.S. wrote, reviewed, and edited the paper; and Y.H. and H.S. acquired the funding and supervised the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Harukazu Suzuki, RIKEN Center for Life Science Technologies, 1-7-22, Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230-0045, Japan; e-mail: harukazu.suzuki@riken.jp.

