

Key Points
DIAPH1-related disorder has a bilineage hematological phenotype of macrothrombocytopenia and neutropenia associated with hearing loss.
Eltrombopag increased proplatelet formation from cultured DIAPH1-related disorder megakaryocytes and improved platelet counts in vivo.
Introduction
The heritable thrombocytopenias (HTs) are genetically heterogeneous rare disorders in which reduced circulating platelet levels may be associated with nonhematological features.1,2 Among recently discovered HTs, DIAPH1-related disorder (D-RD; OMIM #124900) was initially reported in 2 pedigrees with macrothrombocytopenia and hearing loss. This phenotype segregated with a heterozygous p.R1213* variant in DIAPH1, which encodes the cytoskeletal regulator diaphanous homolog 1 (DIAPH1).3 This predicted truncation of the DIAPH1 C terminus diaphanous autoregulatory domain (DAD) and was proposed to confer gain-of-function, resulting in megakaryocyte (MK) cytoskeletal dysregulation and impaired proplatelet formation.3 Macrothrombocytopenia and hearing loss have subsequently been reported in further isolated pedigrees with DAD DIAPH1 variants,4-6 suggesting that D-RD is a distinct syndromic HT. However, other descriptions of similar DIAPH1 variants include hearing loss but not hematological findings.7,8
To provide a full phenotypic description of D-RD and the relationship with different DIAPH1 variants, we report detailed hematological findings from 5 D-RD pedigrees, including the in vitro response and clinical outcome of treatment with the thrombopoietin (TPO) receptor agonist eltrombopag.
Case description
The 5 pedigrees consist of 16 available cases with heterozygous DIAPH1 variants within the DAD (10 males, current ages 2-78 years; Figure 1A). Cases A-2, A-3, A-5, D-7, and E-3 (all with p.R1213*) have already been partially reported by us.3,6 Pedigrees B and C are unreported. Abnormal bleeding was reported in 6 D-RD cases and was predominantly mild and mucocutaneous (Figure 1B). Three D-RD cases had previously received prophylactic platelet transfusions to prevent surgical or obstetric bleeding. The 3 D-RD cases from pedigree E had multiple hospital visits with respiratory tract or cutaneous infections. Bilateral sensorineural hearing loss was detected at neonatal screening or in early childhood in all cases and progressed through childhood. Twelve cases required hearing aids, and 1 case underwent successful cochlear implantation. There were no other consistently reported clinical features.
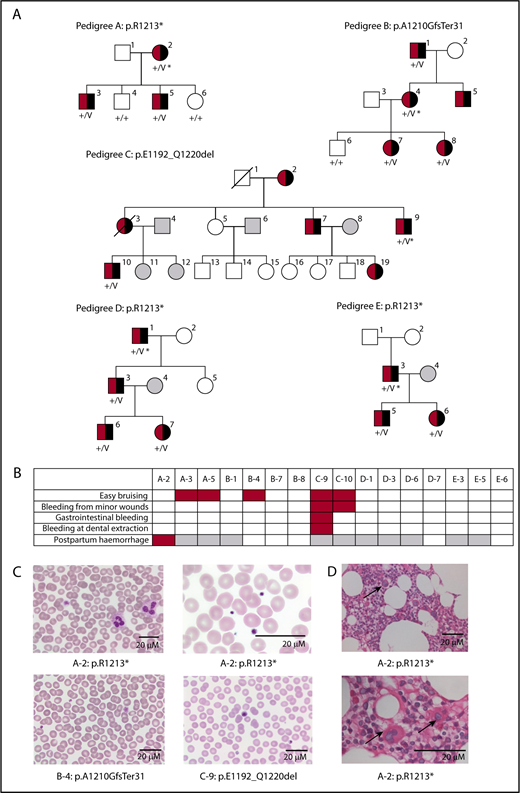
Variants in DIAPH1 associated with D-RD. (A) Pedigree diagrams demonstrating cosegregation of the DIAPH1 variants with sensorineural hearing impairment (black shading) and hematological abnormalities (red shading) in 5 pedigrees. The open symbols indicate unaffected pedigree members. The gray symbols indicate pedigree members with no data available. *Index cases. (B) Annotation of the 16 D-RD cases with Human Phenotype Ontology terms for bleeding symptoms. Red shading indicates the presence of the bleeding symptom. Gray shading indicates that a symptom was not applicable due to patient age or sex. (C) Representative May-Grünwald-Giemsa–stained peripheral blood smears from D-RD cases A-2, B-4, and C-9 representing each of the 3 observed DIAPH1 variants. Original magnification ×40. (D) Hematoxylin and eosin–stained bone marrow biopsy from D-RD case A-2 (R1213*). Granulopoiesis was reduced, with few examples of mature neutrophils. MKs were normal in number but generally small, with hypolobated nuclei (arrows). Original magnification ×100. V, variant DIAPH1 alleles; +, wild-type DIAPH1 alleles.
Variants in DIAPH1 associated with D-RD. (A) Pedigree diagrams demonstrating cosegregation of the DIAPH1 variants with sensorineural hearing impairment (black shading) and hematological abnormalities (red shading) in 5 pedigrees. The open symbols indicate unaffected pedigree members. The gray symbols indicate pedigree members with no data available. *Index cases. (B) Annotation of the 16 D-RD cases with Human Phenotype Ontology terms for bleeding symptoms. Red shading indicates the presence of the bleeding symptom. Gray shading indicates that a symptom was not applicable due to patient age or sex. (C) Representative May-Grünwald-Giemsa–stained peripheral blood smears from D-RD cases A-2, B-4, and C-9 representing each of the 3 observed DIAPH1 variants. Original magnification ×40. (D) Hematoxylin and eosin–stained bone marrow biopsy from D-RD case A-2 (R1213*). Granulopoiesis was reduced, with few examples of mature neutrophils. MKs were normal in number but generally small, with hypolobated nuclei (arrows). Original magnification ×100. V, variant DIAPH1 alleles; +, wild-type DIAPH1 alleles.
Methods
Cases were identified through the National Institute for Health Research BioResource–Rare Diseases (pedigrees A-C; UK REC 13/EE/0325) and Functional and Molecular Characterization of Patients with Inherited Platelet Disorders (pedigrees D-E; Centro Regional de Hemodonación, Universidad de Murcia) programs. Phenotype collection and high-throughput sequencing were as reported previously.6,9,10
Results and discussion
All 16 D-RD cases displayed mild thrombocytopenia on ≥1 occasion (median platelet count, 111 × 109/L; range, 13-209 × 109/L) and enlarged platelets (median mean platelet volume, 12.7 fl; range, 9.3-19.8). Eleven cases also displayed neutropenia on ≥1 occasion (median neutrophil count, 1.33 × 109/L; range, 0.50-4.30) (supplemental Table 1). There were no other morphological abnormalities by light microscopy (Figure 1C). Bone marrow biopsies from cases A-2 and D-3 revealed a normal distribution of cells but reduced granulopoiesis in case A-2. MKs were present in normal numbers but were small with hypolobated nuclei (Figure 1D). Neutrophil adhesion, degranulation, reactive oxygen species generation, and extracellular trap formation were the same in D-RD cases with the p.R1213X and p.A1210GfsTer31 DIAPH1 variants as controls (supplemental Table 2). There were no consistent abnormalities in immunoglobulin concentrations, lymphocyte subset numbers, or lymphocyte proliferation responses (supplemental Table 2). There was no consistent red cell phenotype.
The DIAPH1 variants included the previously reported p.R1213*3,6 and the unreported p.A1210GfsTer31 arising from the dinucleotide deletion DIAPH1 c.3771_3772delAG (pedigree B). Cases from pedigree C harbored an unreported inversion with breakpoints in DIAPH1 introns 26 and 27, predicting in-frame skipping of exon 27 (p.E1192_Q1220del). Consistent with this, a platelet complementary DNA amplicon corresponding to DIAPH1 exons 26-28 was smaller in pedigree C compared with controls and did not contain exon 27 (supplemental Figure 1). All of the DIAPH1 variants predict truncation within the DIAPH1 DAD (Figure 2A), resulting in loss of the RRKR motif and, for p.E1192_Q1220del (pedigree C), also the MDxLLExL motif. These conserved regulatory sequences within the DAD mediate DIAPH1 autoinhibition by competitive binding at the Rho GTPase activation site in the GBD/FH3 domain.11 Cases from pedigree C had more bleeding symptoms and the lowest overall platelet counts in the cohort, suggesting a relationship between the extent of the DAD truncation and phenotype.
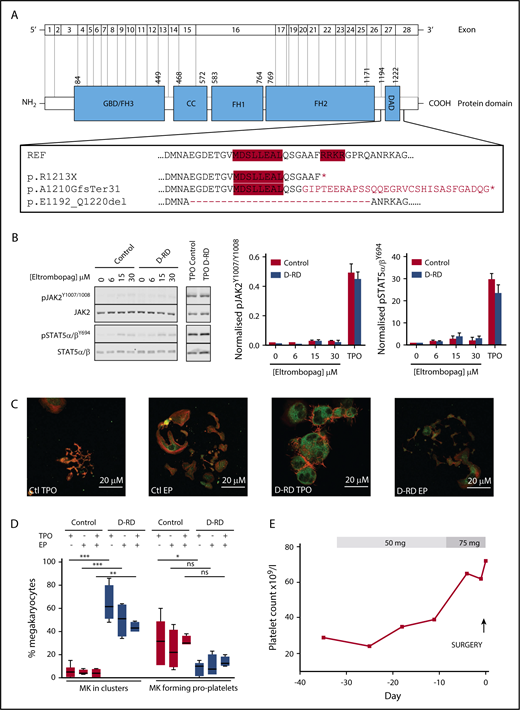
Detailed evaluation of D-RD cases. (A) Schematic representation of DIAPH1 and DIAPH1 protein divided into functional domains, including the DAD near the C terminus. The expanded box shows the wild-type DAD amino acid sequence; the positions of the regulatory RRKR and MDxLLExL sequence motifs are indicated by red shading. The predicted impact of the variants associated with D-RD is shown compared with the reference sequence. The abnormal C terminus amino acid sequence predicted from the p.A1210GfsTer31 variant is indicated in red type. The position of exon 27 residues that are absent with the p.E1192_Q1220del variant is indicated by the dashed line. *Premature stop codon. (B) Representative immunoblot using monoclonal antibodies recognizing p-STAT5α/βY694, total STAT5α/β, pJAK21007/1008, and total JAK2 of lysates from case A-2 and control platelets stimulated with eltrombopag (0-30 μM) or TPO (100 ng/mL) (left panels). Bar graphs of the ratio of phosphorylated/total densitometry signal of 3 D-RD cases (A-2, B-4, and C-9) combined show that eltrombopag causes markedly reduced STAT5α/β and JAK2 phosphorylation compared with TPO and that the extent of phosphorylation in D-RD platelets is the same as controls (middle and right panels). The data are representative of 3 independent experiments expressed as mean ± standard error of the mean. (C) Representative immunofluorescence confocal microscopy images (case A-2 and control) of differentiated peripheral blood–derived CD34+ MKs at day 12 of culture, visualized using anti-integrin β3 (green; CD61) and phalloidin (red; F-actin) staining. In the presence of TPO, D-RD MKs show abnormal clustering, reduced proplatelet formation, and abnormal distribution of F-actin when compared with controls. Reduced proplatelet formation and cluster formation are partially rescued in culture conditions containing eltrombopag (EP). (D) Corresponding bar graphs of aggregate data from duplicate MK-differentiation experiments from 3 unrelated healthy controls and cases A-2, B-4, and C-9, with each of the 3 D-RD variants cultured with TPO (5 μM), EP (3 μM), or TPO (2.5 μM) + EP (2 μM). Data are expressed as mean and standard error of the mean of the percentage of all cultured MKs that associate in clusters and the percentage that are forming proplatelet extensions, as specified in supplemental Methods. (E) Time course of the hematological response to oral eltrombopag administered to case C-9 before elective hip arthroplasty at day 0. Platelet counts were determined using a Sysmex XN analyzer using the fluorescence end point. ***P < .0001, **P < .001, *P < .05, 1-way analysis of variance. ns, not significant.
Detailed evaluation of D-RD cases. (A) Schematic representation of DIAPH1 and DIAPH1 protein divided into functional domains, including the DAD near the C terminus. The expanded box shows the wild-type DAD amino acid sequence; the positions of the regulatory RRKR and MDxLLExL sequence motifs are indicated by red shading. The predicted impact of the variants associated with D-RD is shown compared with the reference sequence. The abnormal C terminus amino acid sequence predicted from the p.A1210GfsTer31 variant is indicated in red type. The position of exon 27 residues that are absent with the p.E1192_Q1220del variant is indicated by the dashed line. *Premature stop codon. (B) Representative immunoblot using monoclonal antibodies recognizing p-STAT5α/βY694, total STAT5α/β, pJAK21007/1008, and total JAK2 of lysates from case A-2 and control platelets stimulated with eltrombopag (0-30 μM) or TPO (100 ng/mL) (left panels). Bar graphs of the ratio of phosphorylated/total densitometry signal of 3 D-RD cases (A-2, B-4, and C-9) combined show that eltrombopag causes markedly reduced STAT5α/β and JAK2 phosphorylation compared with TPO and that the extent of phosphorylation in D-RD platelets is the same as controls (middle and right panels). The data are representative of 3 independent experiments expressed as mean ± standard error of the mean. (C) Representative immunofluorescence confocal microscopy images (case A-2 and control) of differentiated peripheral blood–derived CD34+ MKs at day 12 of culture, visualized using anti-integrin β3 (green; CD61) and phalloidin (red; F-actin) staining. In the presence of TPO, D-RD MKs show abnormal clustering, reduced proplatelet formation, and abnormal distribution of F-actin when compared with controls. Reduced proplatelet formation and cluster formation are partially rescued in culture conditions containing eltrombopag (EP). (D) Corresponding bar graphs of aggregate data from duplicate MK-differentiation experiments from 3 unrelated healthy controls and cases A-2, B-4, and C-9, with each of the 3 D-RD variants cultured with TPO (5 μM), EP (3 μM), or TPO (2.5 μM) + EP (2 μM). Data are expressed as mean and standard error of the mean of the percentage of all cultured MKs that associate in clusters and the percentage that are forming proplatelet extensions, as specified in supplemental Methods. (E) Time course of the hematological response to oral eltrombopag administered to case C-9 before elective hip arthroplasty at day 0. Platelet counts were determined using a Sysmex XN analyzer using the fluorescence end point. ***P < .0001, **P < .001, *P < .05, 1-way analysis of variance. ns, not significant.
To evaluate the safety of the TPO receptor agonist eltrombopag (Novartis, Frimley, United Kingdom) as a potential therapy to increase platelet count in D-RD, we first evaluated its effect on TPO receptor signaling in D-RD platelets. In platelets from healthy controls, eltrombopag activates TPO receptor signaling pathways to a lesser extent than TPO and does not enhance agonist-mediated activation.12 In keeping with this, immune thrombocytopenia patients receiving eltrombopag show no increase in platelet activation in vivo.13 In platelets from D-RD cases with all 3 variants, clinically relevant eltrombopag concentrations stimulated only weak phosphorylation of pJAK2Y1007/1008 and pSTAT5α/βY694 compared with TPO (Figure 2B). This response was similar in healthy controls and indicated no effect of the DIAPH1 variants on the TPO receptor pathway in platelets.
We reported previously that blood cell–derived CD34+ MKs from case A-2 with the DIAPH1 p.R1213* variant displayed abnormal clustering and reduced proplatelet production, as well as abundant and disorganized actin, when cultured with TPO.3 This finding was similar in MKs from cases B-4 (p.A1210GfsTer31) and C-9 (p.E1192_Q1220del) (supplemental Figure 2), supporting a common effect from the different DAD variants (Figure 2C-D). For all 3 cases, there was a trend toward reduced MK clustering and increased proplatelet production after culture of MKs with eltrombopag instead of TPO and further improvement after culture with TPO plus eltrombopag (Figure 2C-D).
We monitored the clinical effect of eltrombopag in case C-9 (DIAPH1 p.E1192_Q1220del) who, unrelated to D-RD, required right hip arthroplasty. However, because of previous platelet transfusions, case C-9 had multiple anti-HLA immunoglobulin G alloantibodies and platelet refractoriness. Eltrombopag (50 mg) was administered once daily from day −29 before surgery, which was increased to 75 mg once daily from day −9 until day −1. The platelet count, which was determined using the PLT-F detection end point (XN-Series analyzer; Sysmex, Kobe, Japan), increased from a baseline of 29 × 109/L to 72 × 109/L on day −1 before surgery (Figure 2E). The patient also received tranexamic acid for 72 hours from the start of surgery but not platelet transfusion. Hemostasis was satisfactory (total estimated blood loss 500 mL vs 493 mL for controls undergoing similar surgery14 ), and there were no adverse events.
This largest reported series of 16 cases illustrates that D-RD is a dominant disorder characterized by macrothrombocytopenia, neutropenia, and hearing loss. This analysis revealed no defects in neutrophil function, and no lymphoid or red cell abnormalities. Although neutropenia potentially accounted for the recurrent infections observed in 1 D-RD pedigree (E), clinical immunodeficiency was absent in the other cases. No cases had renal disease, cataracts, or neutrophil inclusions, distinguishing D-RD from MYH9-related disorder in which there may also be macrothrombocytopenia and hearing loss.15-17 The D-RD phenotype was associated with chain truncation variants close to the DIAPH1 C terminus, resulting in loss of conserved regions responsible for autoinhibitory interactions within the DIAPH1 protein.11 This suggests a distinct molecular pathogenesis for D-RD, arising from a gain-of-function effect.
This report also illustrates that eltrombopag partly rescues defective proplatelet formation in D-RD MKs cultured in vitro but that TPO receptor signaling responses in D-RD platelets are unaltered. We provide proof of concept that, similar to in MYH9-related disorder,18,19 short-term eltrombopag may allow temporary correction of platelet counts in D-RD cases before surgery, enabling avoidance of platelet transfusion.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Ingeborg Hers, Verónica Palma-Barqueros, and Nerea Mota for assistance with some experiments. This study used data generated by the National Institute for Health Research (NIHR) BioResource.
NIHR BioResource–Rare Diseases is funded by the NIHR (award number RG65966). The NIHR BioResource projects were approved by research ethics committees in the United Kingdom and appropriate national ethics authorities in non-UK enrollment centers. M.L.L. and J.R. are members of the Inherited Platelet Disorders Project, Hemorrhagic Diathesis Working Group, Spanish Foundation for Hemostasis and Thrombosis. S.K.W. is supported by a Medical Research Council Clinical Research Training Fellowship (MR/K023489/1). K.D. is supported as an HSST trainee by NHS Health Education England. S.F.M. is supported by the British Heart Foundation (PG/16/3/31833). N.V.M. is supported by the British Heart Foundation (PG/13/36/30275). M.L.L. and J.R. are supported by grants from Instituto de Salud Carlos III and Feder (PI17/01311 and CB15/00055), Fundación Séneca (19873/GERM/15), and Fundación Española de Trombosis y Hemostasia. K.F. is supported by the Fund for Scientific Research-Flanders (FWO-Vlaanderen, Belgium, G.0B17.13N) and the Research Council of the University of Leuven (BOF KU Leuven, Belgium, OT/14/098). W.H.O. is supported by the British Heart Foundation, the European Commission, the Medical Research Council, NIHR, the Wellcome Trust, and the National Health Service Blood and Transplant. A.D.M., S.K.W., C.B., and S.F.M. are supported by the Bristol NIHR Biomedical Research Centre.
The views expressed in this publication are those of the authors and not necessarily those of the National Health Service, the National Institute for Health Research, or the Department of Health.
Authorship
Contribution: S.K.W. and A.D.M. wrote the manuscript with assistance from K.D., K.F., and J.R.; C.B., M.L.L., S.G.O., T.S., and C.H.T. provided samples and clinical data; W.N.E. provided bone marrow and blood smear analyses; N.V.M. provided DNA sequencing and analysis for pedigree E; C.K., S.F.M., C.T., and S.K.W. performed laboratory experiments and analyzed data; K.D. and K.G. managed and chaired the ThromboGenomics program, respectively; S.P. coordinated the NIHR BioResource–Rare Diseases Bleeding and Platelet Disorders project, including ethics and governance; and K.F., M.A.L., W.H.O., and A.D.M. contributed to the study design.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the NIHR BioResource–Rare Diseases Consortium appears in the online appendix and can be accessed at https://bioresource.nihr.ac.uk/researchers/researchers/acknowledgement/.
Correspondence: Andrew D. Mumford, Research Floor 7, Bristol Royal Infirmary, University of Bristol, Bristol BS2 8HW, United Kingdom; e-mail: a.mumford@bristol.ac.uk.

