

Key Points
Mesenchymal stem cells provide a potent human stem cell niche for development of mature NK cells from hematopoietic stem cells.
Expression of KIR occurs independently of the presence of HLA class I ligands in the bone marrow niche model.

Abstract
The development of mature natural killer (NK) cells expressing killer cell immunoglobulin-like receptors (KIRs) depends on cell contact–dependent signals from nonhematopoietic cells. So far, detailed studies of this process have been hampered by the lack of an appropriate in vitro model. Here, human bone marrow–derived mesenchymal stem cells (MSCs), generated under good manufacturing practice (GMP) conditions, are established as a supportive niche for in vitro NK cell differentiation. In the presence of MSCs, cord blood and bone marrow–derived hematopoietic stem and progenitor cells (HSPCs) effectively and reproducibly differentiated into mature KIR-expressing NK cells. Notably, the novel in vitro differentiation assay enabled us to analyze the impact of HLA class I ligands on KIR repertoire development. To this end, a panel of MSC lines divergent for expression of the major KIR ligands C1, C2, and Bw4 was used for NK cell differentiation. The resulting NK cell repertoires were independent of the presence of specific KIR ligands on MSCs and were, in fact, invariably dominated by expression of the C1-specific inhibitory KIR2DL3. Similarly, short hairpin RNA–mediated knockdown of HLA class I ligands on MSCs did not delay or change the course of KIR expression. Our data suggest that the initial acquisition of KIRs during NK cell development is biased toward recognition of C1 ligands, irrespective of the presence of self-ligands. Altogether, the MSC/HSPC model constitutes a novel platform to study NK cell development in a human stem cell niche. Moreover, the system constitutes a promising GMP-compliant platform to develop clinical-grade NK cell products from cord blood HSPCs.
Introduction
Killer cell immunoglobulin-like receptors (KIRs) are expressed on natural killer (NK) cells and constitute sensors for HLA class I expression.1 Only those NK cells that express a suitable inhibitory KIR are able to detect downregulation of a given HLA class I allotype on a target cell. Analysis of NK cell development in vitro, as well as following hematopoietic stem cell transplantation, showed that the formation of NK cell repertoires is a sequential process that starts with acquisition of HLA-E–specific NKG2A as the first HLA class I–specific receptor. In the next step, a subset of NK cells acquires HLA class I–specific KIRs, starting with the HLA-C1–specific inhibitory receptor KIR2DL2 or KIR2DL3 (depending on the given KIR genotype).2,3 Subsequently, KIR repertoires are diversified through clonal expression of other inhibitory and stimulatory KIRs. Notably, although the different steps of receptor acquisition are partially overlapping, a consistent observation in vitro and in vivo is that expression of the HLA-C2–specific KIR2DL1 is delayed, even in individuals possessing no C1 ligands.2,4 Nonetheless, analysis of NK cell repertoires in cord blood (CB) revealed that the frequency of KIR2DL1-expressing NK cells is comparable to KIR2DL3, suggesting that, by the time of birth, the naive NK cell repertoire is no longer biased toward C1-specific KIR expression.5
It is still unclear how KIR repertoires are actually adjusted to HLA class I ligands in a given individual. It was previously shown that NK cells expressing KIRs for self-ligands are found at elevated frequencies in healthy adults.6,7 However, no adjustment of self-specific NK cells was found in neonatal blood, suggesting that the enrichment of self-specific KIR in adults is primarily driven by the immunological history, such as virus infections.5 This idea is supported by studies showing that expansions of NK cells with self-specific KIRs were particularly prominent in human cytomegalovirus–infected individuals, who coexpressed the HLA-E–specific stimulatory receptor NKG2C.8 How other infectious agents influence NK cell repertoires remains unclear.
Differentiation of NK cells from hematopoietic stem and progenitor cells (HSPCs) in vitro constitutes an important experimental tool to improve our understanding of the factors that regulate the formation of NK cell repertoires. Although NK cells can be generated by culture of HSPCs in stroma cell–free conditions, purely cytokine-mediated stimuli are not sufficient to induce KIR expression, possibly due to a lack of signals promoting the generation of more mature NK cells.9 In contrast, mature KIR-expressing NK cells can be generated more efficiently in stroma cell–based NK cell–differentiation assays. So far, these in vitro systems are based on coculture of human HSPCs with xenogeneic murine stroma cells.10 However, in these models, mechanistic studies of NK cell differentiation are limited to those factors that are conserved between mouse and humans, 2 species that are separated for roughly 60 million years. In particular, the absence of HLA class I in rodent stroma cells hampers detailed analysis of ligand-induced KIR regulation.
Mesenchymal stem cells (MSCs) are multipotent mesenchymal stromal cells that are able to differentiate into a variety of mesodermal cell types, such as osteoblasts, chondrocytes, and adipocytes.11 After birth, MSCs primarily reside in the bone marrow (BM) but are also found at extramedullary sites, such as CB, fat, and many other tissues. In addition to their function as multipotent stromal progenitor cells, MSCs have well-described immunoregulatory capabilities, which include being able to inhibit T cell responses, as well as modify dendritic cell differentiation and function.12 Importantly, MSCs are also an integral part of the hematopoietic stem cell niche in BM, regulating homing and controlling the fate of HSPCs.13 In the present study, based on their described supportive functions on hematopoiesis, MSCs were evaluated as an in vitro surrogate of a human hematopoietic niche for NK cell differentiation. MSC lines generated from the BM of young children robustly supported the development of mature KIR-expressing and functionally competent NK cells and enabled us to systematically address the role of HLA class I ligands in shaping the human NK cell repertoire.
Materials and methods
Cell lines and cell culture
For isolation and expansion of human GMP-grade MSCs, 5 to 20 mL of heparinized human BM aspirate was obtained from healthy volunteer donors after informed consent. A detailed description of MSC isolation and cultivation is included in supplemental Methods. This study was approved by the ethics committee of the Medical Faculty of Heinrich-Heine-University (no. 1830 and 3880). Unrestricted somatic stem cells (USSCs) were isolated from human umbilical CB and cultivated as described previously.14 The murine embryonic liver stroma cell line EL08.1D2 was kindly provided by R. Oostendorp and was cultured in Iscove modified Dulbecco medium (Lonza) with 15% fetal calf serum, 5% horse serum, 1% penicillin/streptomycin, and 400 μM 2-mercaptoethanol at 33°C. The murine BM stroma cell lines OP9/OP9-DL1 were cultured in high-glucose Dulbecco’s modified Eagle medium with 1% l-glutamine, 10% fetal calf serum, and 50 μM 2-mercaptoethanol. The fibroblast cell line STF5, generated from a healthy donor, was kindly provided by J. Zimmer (CRP-Santé, Strassen, Luxembourg).
Analysis of NK cell function
NK cells harvested from MSC/HSPC cultures were mixed with K562 target cells at an effector/target ratio of 1:1. The CD107 mobilization assay was performed as previously described.15 For intracellular measurement of intracytoplasmic interferon-γ, effector and K562 target cells were coincubated for 6 hours. After the first hour, Brefeldin A (Sigma-Aldrich, St. Louis, MO) was added at a concentration of 10 µg/mL. Cells were fixed and permeabilized with Cytofix/Cytoperm solution (BD Biosciences) and stained intracellularly with anti–IFN-γ monoclonal antibody. For determination of cytotoxicity, K562 target cells were stained with CFDA SE (Vybrant CFDA SE Cell Tracer Kit; Invitrogen, Carlsbad, CA) as described previously.15
shRNAs and lentiviral transduction
Knockdown of β-2-microglobulin (β2M) was performed using previously described short hairpin RNAs (shRNAs).16 A detailed description of the shRNA cloning strategy and virus production is found in supplemental Methods.
KIR and HLA class I genotyping
KIR genotyping was carried out by single specific primer–polymerase chain reaction, as previously described.17 HLA class I typing was performed by Luminex technology (One Lambda). Confirmatory typing for C1/C2 epitopes was performed by single specific primer–polymerase chain reaction, as previously described.18
Statistics
All tests were performed at the 2-sided 0.05 significance level, as indicated. All statistical analyses were performed using GraphPad Prism software.
Results
Human MSCs support NK cell development from hematopoietic progenitor cells
To establish an in vitro model to analyze the impact of HLA class I expression on NK cell development and formation of KIR repertoires, we aimed at identifying a suitable human cell source that enables efficient generation of NK cells from HSPCs. To this end, we used a panel of MSC lines that were established from BM of children (age 1.5-8.5 years) under GMP conditions. In addition, we used MSC lines generated from umbilical CB (USSCs), as described previously.14 These MSC lines were compared with human fibroblasts (STF5). HSPCs were isolated from CB and seeded on the respective irradiated stromal monolayers. Culture conditions and cytokines were adapted from previous studies using murine feeder cells.10 As shown in Figure 1, all MSC lines supported NK cell differentiation from CB HSPCs. Starting with 3000 HSPCs, 2 to 4 million NK cells were generated at the end of the sixth week of culture, translating to a >1000-fold cell-expansion rate (Figure 1A-B). In contrast, coculture of HSPCs with fibroblasts did not support NK cell development at all. Notably, the frequency of NK cells expressing HLA class I–specific NK cell receptors (CD94:NKG2A and KIR) was highest in the presence of BM-derived MSCs (Figure 1C-D). CB-derived USSC lines supported NK cell receptor expression as well, but to a lesser extent (P < .001 for NKG2A and KIR, Mann-Whitney U test). Based on these observations, BM-derived MSCs were used as supportive feeder cells in subsequent experiments. In a typical experiment, an expansion phase of HSPCs in the first week was followed by disappearance of the stem cell marker CD34 and upregulation of the activation and differentiation marker CD38 in the second week (data not shown). By day 28, ∼50% of the CD56+ cells expressed the NK cell marker NKG2A and coexpressed inhibitory KIR2DL1, KIR2DL3, and KIR3DL1, which are characteristic features of fully mature NK cells (Figure 1E). No expression of CD3 was found at any point during the culture with MSCs (data not shown). Analysis of stage-specific markers of NK cell development revealed the accumulation of CD94+ cells, as well as the acquisition of CD16 as a marker of functional maturation, which was found on ∼10% of NK cells. CD57 as a marker of late-stage NK cells remained low at all analyzed time points (supplemental Figure 1). We also looked at the degranulation activity of NK cells developed on MSCs. The most frequent subset, NKG2A+KIR− NK cells, exhibited significantly lower degranulation against K562 than the next most frequent subset (NKG2A+KIR2DL3+) (supplemental Figure 2). These data are in line with functional data of primary NK cells showing additive effects of multiple licensed receptors19 and suggest a licensing effect of KIR expression in this system. For other subsets, such as single KIR+ NK cells, CD107 mobilization could not be reliably determined because of the low frequencies.
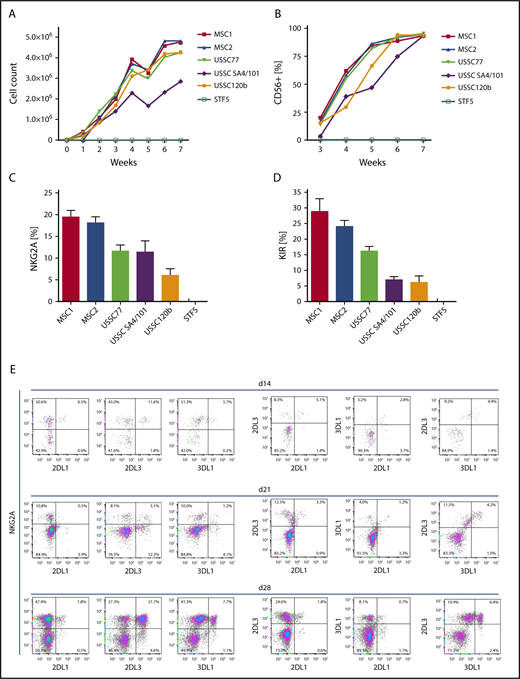
BM-derived MSCs efficiently support the generation of KIR-expressing NK cells from early hematopoietic progenitors. CD34+ HSPCs were isolated from CB and seeded at 3 × 103 cells per well on monolayers of the indicated stromal cell types (MSC, USSC) or fibroblasts (STF5). Cell number (A) and expression of CD56 (B) were measured weekly during culture by flow cytometry. Maximum expression frequency of NKG2A (C) and KIR (D) was monitored for each of the indicated cell lines using a mixture of KIR-specific monoclonal antibodies for inhibitory KIR2D and KIR3D receptors (data are mean frequency and standard deviation of 3 independent experiments). (E) Flow cytometric analysis of cultures at day 14 (top panel), day 21 (middle panel), and day 28 (bottom panel). Cells were gated on the CD56+ subset, and expression of the NK cell receptors KIR2DL1, KIR2DL3, KIR3DL1, and NKG2A was analyzed.
BM-derived MSCs efficiently support the generation of KIR-expressing NK cells from early hematopoietic progenitors. CD34+ HSPCs were isolated from CB and seeded at 3 × 103 cells per well on monolayers of the indicated stromal cell types (MSC, USSC) or fibroblasts (STF5). Cell number (A) and expression of CD56 (B) were measured weekly during culture by flow cytometry. Maximum expression frequency of NKG2A (C) and KIR (D) was monitored for each of the indicated cell lines using a mixture of KIR-specific monoclonal antibodies for inhibitory KIR2D and KIR3D receptors (data are mean frequency and standard deviation of 3 independent experiments). (E) Flow cytometric analysis of cultures at day 14 (top panel), day 21 (middle panel), and day 28 (bottom panel). Cells were gated on the CD56+ subset, and expression of the NK cell receptors KIR2DL1, KIR2DL3, KIR3DL1, and NKG2A was analyzed.
We next wanted to determine how human MSC-supported NK cell development compares with established protocols that are based on murine stroma cells. So far, most studies used the murine fetal hepatic stroma cell line EL08.1D2 or the murine BM stroma cell line OP9, the latter often as derivatives expressing Notch ligands, such as Delta-like-1 (OP9-DL1). To this end, we cocultured CB-derived HSPCs on these 3 murine stroma cell lines in parallel with human MSCs. Generally, EL08.1D2 and MSCs generated the highest frequency of KIR2DL3, which is the first KIR appearing at the surface during NK cell development. OP9-DL1 and the parental OP9 line generated fewer and no KIR2DL3-expressing NK cells, respectively (Figure 2A-B). With regard to NK cell effector functions, cytotoxicity against K562 target cells was equivalent between EL08.1D2 and MSCs (Figure 2C). Similarly, intracellular expression of the cytotoxic effector molecules perforin and granzyme B were comparable (Figure 2D).

KIR expression and functional analysis of NK cells developed on human MSCs and murine stromal feeder cells. CD34+ HSPCs were isolated from CB and seeded onto human MSCs and 3 murine stromal feeder cells (EL08.1D2, OP9, and OP9-DL1). (A) Representative flow cytometric analysis of KIR2DL3 expression on NK cells on day 25. (B) Frequency of KIR2DL3 expression (data are mean and standard deviation of 3 independent experiments). (C) Comparison of granzyme B and perforin expression in NK cells developed on EL08.1D2 or MSCs by intracytoplasmic staining and flow cytometry. (D) Cytotoxicity of NK cells cultured on EL08.1D2 or MSCs against K562 target cells at an effector/target ratio of 5:1. *P < .05, Fisher t test.
KIR expression and functional analysis of NK cells developed on human MSCs and murine stromal feeder cells. CD34+ HSPCs were isolated from CB and seeded onto human MSCs and 3 murine stromal feeder cells (EL08.1D2, OP9, and OP9-DL1). (A) Representative flow cytometric analysis of KIR2DL3 expression on NK cells on day 25. (B) Frequency of KIR2DL3 expression (data are mean and standard deviation of 3 independent experiments). (C) Comparison of granzyme B and perforin expression in NK cells developed on EL08.1D2 or MSCs by intracytoplasmic staining and flow cytometry. (D) Cytotoxicity of NK cells cultured on EL08.1D2 or MSCs against K562 target cells at an effector/target ratio of 5:1. *P < .05, Fisher t test.
MSCs express KIR ligands on the cell surface and are tolerated by NK cells
Our finding that human MSCs provide a novel supportive stem cell niche for NK cell development opened the opportunity to analyze the regulation of HLA class I–specific NK receptor expression. To serve as a suitable model, it was important to check whether irradiated MSCs remained present throughout the coculture experiment and whether they would express the appropriate HLA class I ligands. As shown in Figure 3, MSCs were readily detectable and remained viable for ≥4 weeks following start of the culture. Most of the MSCs were found in close contact with developing lymphocytes, presumably NK cells (Figure 3A). Thus, whereas human MSCs were unaffected by the effector NK cells generated in the culture, murine EL08 stroma cells disappeared after 2 to 3 weeks of culture, coinciding with the appearance of mature NK cells (data not shown). The results are in line with the observation that peripheral blood mononuclear cell-derived NK cells exerted less cytotoxicity against MSCs than against EL08, although this was not statistically significant (supplemental Figure 3). Lack of reactivity of MSCs against NK cells does not seem to be due to a lack of ligands for stimulatory NK cell receptors. Among others, MSCs expressed the NKG2D ligands ULBP1-4 and MIC-A, as well as the NKp30 ligand B7-H6 (supplemental Figure 4), but not the checkpoint ligands PD-L1, CD80, or CD86 (data not shown).

Expression of HLA class I on the cell surface of MSCs during in vitro NK cell development. (A) Representative micrographs (original magnification ×100)depicting the morphology of MSCs during NK cell differentiation from day 0 to day 28. MSCs keep their typical extended shape and are found in close contact with developing NK cells. (B) HLA class I expression on the surface of MSCs at the start and after 2 and 4 weeks of culture using the pan–HLA class I antibody W6/32. Gating was performed on the basis of a control staining with secondary antibody only. (C) Expression of HLA-C on MSCs at day 14 using isotype control (left panel) and FITC-conjugated HLA-C–specific monoclonal antibody DT9 (right panel).
Expression of HLA class I on the cell surface of MSCs during in vitro NK cell development. (A) Representative micrographs (original magnification ×100)depicting the morphology of MSCs during NK cell differentiation from day 0 to day 28. MSCs keep their typical extended shape and are found in close contact with developing NK cells. (B) HLA class I expression on the surface of MSCs at the start and after 2 and 4 weeks of culture using the pan–HLA class I antibody W6/32. Gating was performed on the basis of a control staining with secondary antibody only. (C) Expression of HLA-C on MSCs at day 14 using isotype control (left panel) and FITC-conjugated HLA-C–specific monoclonal antibody DT9 (right panel).
With regard to HLA class I expression, flow cytometric analysis of MSCs with a pan–HLA class I–specific reagent (W6/32) demonstrated strong expression on the cell surface for ≥28 days following irradiation (Figure 3B). Next, an HLA-C–specific antibody (DT9) was used to determine whether MSCs would also express HLA-C molecules constituting the ligands for inhibitory KIR2D receptors. For this purpose, MSCs were trypsinized and purified from cocultures after 2 weeks by flow cytometric cell sorting. As seen in Figure 3C, the majority of MSCs (>75%) still displayed substantial amounts of HLA-C on the cell surface.
Expression of KIR during NK cell differentiation is independent of HLA class I ligands
The above data suggest that human BM-derived MSCs fulfill all of the basic requirements of a suitable model to evaluate the role of HLA class I ligands in the formation of NK cell repertoires. In the basic experimental setup, HSPCs were isolated from CB and seeded on a panel of MSC lines divergent for the major KIR ligands HLA-C1, HLA-C2, and Bw4, as well as on murine EL08 stroma cells. CB units were preselected for homozygosity of group A KIR haplotypes (A/A genotype). The A/A KIR genotype ensured identical KIR gene dose, including the presence of 2 copies of all 3 major inhibitory receptors (KIR2DL1, KIR2DL3, and KIR3DL1), thus minimizing the influence of KIR gene polymorphism between experiments. Moreover, A/A genotypes do not encode stimulatory KIR that could cross-react with the KIR-specific antibody reagents, thereby facilitating interpretation of the flow cytometric results.20
As shown in Figure 4, the first KIR+ NK cells are typically detected in the third week of culture and express KIR2DL3, followed by expression of KIR2DL1 (Figure 4A-B), as well as other KIRs in the fourth week (supplemental Figure 5). The kinetics of KIR expression was quite similar and independent of the HLA-C–encoded KIR ligands. In general, KIR2DL3 expression was significantly stronger (10%-20% of CD56+ cells) than that of KIR2DL1 (1-6% of CD56+ cells), even in MSC lines that were homozygous for C2 ligands and, thus, missing C1 ligands. Moreover, the frequency of KIR2DL1 was not increased in coculture with C2/C2 MSCs compared with C1/C1 MSCs (Figure 4B). Similarly, KIR3DL1 expression was not influenced by the presence of Bw4 ligands on MSCs (supplemental Figure 3). These observations contrast with previous data from population studies showing increased frequencies of inhibitory KIRs in the presence of the respective ligands.6,7 In a second set of experiments, HSPCs from 3 different CBs, which were preselected for presence of the A/A KIR genotype, were differentiated on the indicated MSC lines (Figure 4C). The KIR2DL1/KIR2DL3 ratio always remained <0.3, and it did not correlate with the HLA-C ligands presented by the MSCs (Figure 4C). Again, this is in contrast to a previous population study demonstrating that the KIR2DL1/KIR2DL3 ratio is well below 1 in C1/C1 adults and >1 in C2/C2 adults.7
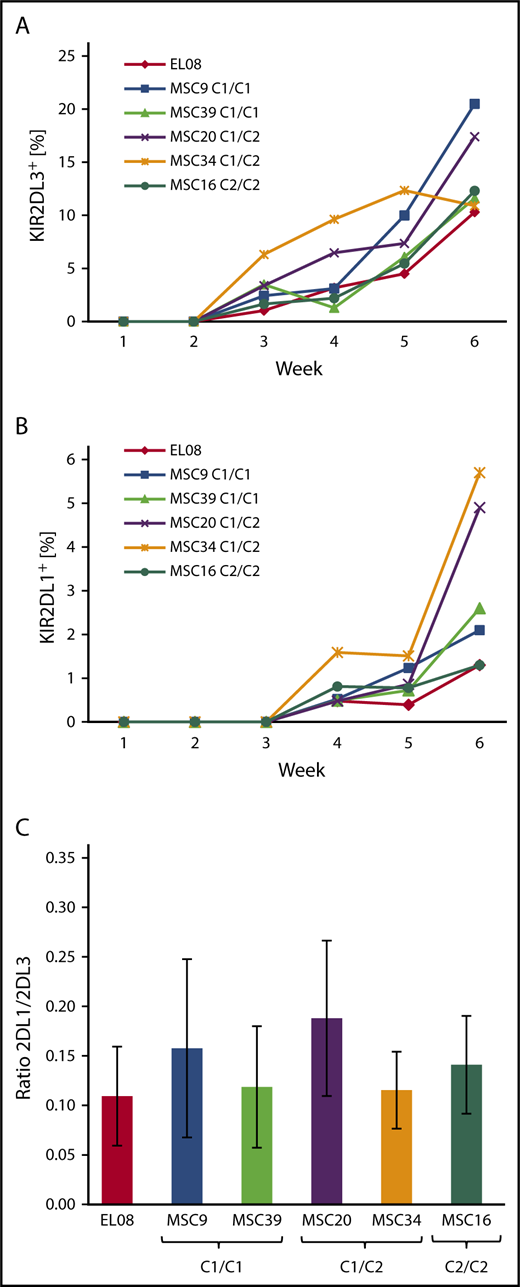
KIR repertoires do not adapt to stromal HLA class I ligands during NK cell development. Frequency of KIR2DL3 (A) and KIR2DL1 (B) expression during the course of NK cell differentiation of HSPCs from a single CB with KIR A/A genotype on a panel of 5 MSC lines with divergent HLA-C ligands and EL08.1D2. (C) KIR2DL1/KIR2DL3 ratios (mean and standard deviation) of NK cells at week 5 of differentiation of CB-derived HSPCs (n = 3) on EL08.1D2 and the indicated MSC lines. All CB had an A/A KIR genotype. Differences between MSC cultures were nonsignificant by the Fisher t test.
KIR repertoires do not adapt to stromal HLA class I ligands during NK cell development. Frequency of KIR2DL3 (A) and KIR2DL1 (B) expression during the course of NK cell differentiation of HSPCs from a single CB with KIR A/A genotype on a panel of 5 MSC lines with divergent HLA-C ligands and EL08.1D2. (C) KIR2DL1/KIR2DL3 ratios (mean and standard deviation) of NK cells at week 5 of differentiation of CB-derived HSPCs (n = 3) on EL08.1D2 and the indicated MSC lines. All CB had an A/A KIR genotype. Differences between MSC cultures were nonsignificant by the Fisher t test.
As shown above, MSCs remain present and express HLA class I ligands throughout the culture period. Nonetheless, it still could not be excluded that other factors that are essential for proper NK cell repertoire formation, such as cytokine production or expression of stimulatory ligands, fade over time on the MSC lines. Thus, we performed replating experiments in which developing NK cells were removed from the coculture with MSCs after 14 days and seeded on fresh MSC layers. The presence of fresh MSCs did not change the general picture that KIR2DL3 was the dominant KIR throughout the culture (supplemental Figure 6). The ratio of KIR2DL3/KIR2DL1 expression remained low, independent of the expression of C1 or C2 ligands on MSCs.
The previous experiments had shown that NK cell development from CB-derived HSPCs was not significantly influenced by HLA class I ligands; however, the common view is that early progenitors with NK cell potential primarily reside in the BM. Thus, we wanted to determine whether BM-derived HSPCs respond differently to the presence of HLA class I ligands. To this end, HSPC samples with A/A KIR genotype were isolated from adult BM and cocultured with MSC lines, similar to the experiments described for CB-derived HSPCs (Figure 5). In general, KIR expression was less efficient from BM-derived HSPCs compared with CB. Nonetheless, KIR repertoires were again dominated by KIR2DL3 expression (Figure 5A-B), regardless of whether they developed on HLA-C1– or HLA-C2–expressing MSC lines. The expression of KIR2DL1 (Figure 5A) and KIR3DL1 (Figure 5C) remained low throughout the culture and did not increase in the presence of C2 or Bw4 ligands, respectively.

Lack of adaptation to stromal HLA class I ligands is independent of the HSPC source. HSPCs from adult BM were differentiated to NK cells on 2 MSC lines with divergent HLA-C–encoded KIR ligands. At the indicated time points, the frequency of KIR2DL1 and KIR2DL3 (A), the ratio of KIR2DL1/KIR2DL3 (B), and the frequency of KIR3DL1 (C) were calculated (data are mean and standard deviation of 3 independent experiments).
Lack of adaptation to stromal HLA class I ligands is independent of the HSPC source. HSPCs from adult BM were differentiated to NK cells on 2 MSC lines with divergent HLA-C–encoded KIR ligands. At the indicated time points, the frequency of KIR2DL1 and KIR2DL3 (A), the ratio of KIR2DL1/KIR2DL3 (B), and the frequency of KIR3DL1 (C) were calculated (data are mean and standard deviation of 3 independent experiments).
Knockdown of MHC class I on MSCs does not impair NK cell development or KIR expression
The previous experiments had clearly shown that the formation of KIR repertoires during NK cell development in vitro is not influenced by the presence of HLA class I–encoded KIR ligands. In a further experiment, we wanted to determine whether the complete absence of HLA class I molecules, including classical HLA-A, HLA-B, and HLA-C and the nonclassical HLA-E on stroma cells, would change the course of NK cell development and KIR repertoire formation. To this end, MSCs were lentivirally transfected with shRNA against β2M. The invariant β2M chain constitutes an essential part of the HLA class I molecule, and silencing of β2M transcription leads to downregulation of HLA class I expression on the cell surface. The shRNA leading to the strongest reduction in β2M mRNA levels was chosen, leading to complete downregulation of β2M in 80% of MSCs (supplemental Figure 7). MSCs with no detectable HLA class I expression were purified by cell sorting and used as supportive cells for NK cell differentiation.
In general, the process of NK cell development on HLA class I–deficient MSC feeder cells proceeded normally; in each differentiation stage, the relevant surface markers, such as CD56 and CD94:NKG2A, were expressed similar to controls (Figure 6A; data not shown). With regard to KIR expression, no changes in the kinetics of expression or the type of KIR were detected (Figure 6B-D). Similar to previous experiments, HLA-C1–specific KIR2DL3 was the most frequent KIR at early time points, followed by KIR3DL1, which was most strongly upregulated between weeks 4 and 6. Expression of HLA-C2–specific KIR2DL1, although increasing over time, did not go above 1% of NK cells, irrespective of the presence of HLA-C2 on the supporting MSC line.

Knockdown of HLA class I expression on MSCs does not change the course of KIR repertoire development. HSPCs from CB were differentiated on MSCs using shRNA-mediated β2M knockdown (B2M), empty vector control (empty), or no treatment (wt). The development of CD56+ NK cells (A) and the expression of KIR2DL1 (B), KIR2DL3 (C), and KIR3DL1 (D) were monitored by flow cytometry at the indicated time points. Mean values and standard deviations of 3 independent experiments are shown.
Knockdown of HLA class I expression on MSCs does not change the course of KIR repertoire development. HSPCs from CB were differentiated on MSCs using shRNA-mediated β2M knockdown (B2M), empty vector control (empty), or no treatment (wt). The development of CD56+ NK cells (A) and the expression of KIR2DL1 (B), KIR2DL3 (C), and KIR3DL1 (D) were monitored by flow cytometry at the indicated time points. Mean values and standard deviations of 3 independent experiments are shown.
Discussion
The present study introduces a model of human NK cell development that enables efficient generation of mature and fully functional NK cells from HSPCs in coculture with human MSCs. In vitro–generated NK cells had the characteristic mature phenotype CD3−CD56+NKG2A+KIR+, expressed perforin and granzyme, and exhibited cytotoxicity against HLA class I− target cells. The supportive MSC lines were established in GMP-grade conditions without xenogeneic components and are routinely used as cellular therapeutics in the clinic. Thus, in addition to being useful to address basic questions of human NK cell development, the HSPC/MSC coculture system provides a promising platform for applications aiming at producing clinical-grade NK cell products.
MSCs have pleiotropic functions as stem cells of bone, cartilage, and fat cells and, furthermore, play diverse roles in hematopoiesis and in the regulation of immune effector functions; as part of the stem cell–supporting microenvironment of the BM niche, MSCs promote differentiation of early HSPCs toward myeloid and lymphoid lineages, including NK cells.21 In general, MSCs constitute a useful novel tool for mechanistic studies of the role of stroma cell factors in NK cell development. Factors thought to contribute in trans to NK cell development, such as HLA class I, are easily knocked down or overexpressed in MSCs. Importantly, the MSC lines remained present in the cocultures until the late stages of NK cell development, and it was neither necessary nor beneficial to replate NK cell cultures to fresh MSC layers. In contrast, murine stroma cell layers were disappearing in coculture with HSPCs by the time that the first NK cells were observed, after 2 to 3 weeks of culture. As reported previously, it is likely that effector NK cells developing in the cultures quickly kill the xenogeneic feeder cells,22 whereas they do not seem to attack MSCs. This phenomenon is probably not due to HLA class I–mediated protection, because MSCs with knocked-down HLA class I were still not killed by the developing NK cells.
The efficient support of NK cell development, the expression of HLA class I, including HLA-C–encoded KIR ligands, and the sustained presence in culture render MSCs a suitable model to study the influence of HLA class I on the formation of NK cell repertoires. It was previously shown that the dimorphic HLA-C–encoded KIR ligands, the HLA-C1 and HLA-C2 epitopes, are associated with expansion of NK cells recognizing the respective ligands, such that C2/C2 individuals had more NK cells expressing KIR2DL1, and C1/C1 individuals had more NK cells expressing KIR2DL2/3.7 A similar increase was also seen for KIR3DL1 in individuals having the Bw4 ligand.6 Notably, a similar adaptation of the NK cell repertoire to HLA class I was not seen in neonatal blood.5 Thus, it remained unclear whether the missing adaptation seen in CB samples was a phenomenon restricted to neonatal blood, a source with many immunosuppressive features, or, in general, NK cells adapt to HLA class I ligands only later in life (eg, in response to pathogenic challenges). The experiments presented in this study clearly argue against a major role for HLA class I ligands in the formation of KIR repertoires during NK cell development and favor a model in which KIR expression is initially determined primarily by genetic polymorphism and only later adapts to its autologous HLA class I–encoded ligands as part of the individual’s immunological history. In the present experiments, expression of C1, C2, and Bw4 ligands did not lead to any detectable adaptation of KIR expression. Several MSC lines having the same KIR ligands were used in the experiments to exclude chance variation in the supportive capacity of individual MSC lines. In fact, in all conditions, developing NK cell repertoires followed a sequential order of receptor acquisition, starting with CD94:NKG2A, followed by the C1-specific KIR2DL2/3, and then later acquisition of the Bw4-specific KIR3DL1 and the C2-specific KIR2DL1, the latter always with the lowest frequency. These observations were also independent of whether CB or BM was the source of HSPCs. Another possibility to consider is the adaptation of cognate KIR to HLA class I in cis, which was described for murine Ly49 receptors.23 CB samples of divergent HLA class I types all followed a similar order of NK receptor expression, although the magnitude varied among different CB samples. Moreover, the expression of C1 or C2 ligands on CB did not play any detectable role in KIR expression when using murine EL08 stroma cells (supplemental Figure 8; supplemental Table 1). These observations make a cis-dependent adaptation process for KIRs unlikely. The above observations are compatible with studies analyzing NK cell reconstitution following clinical hematopoietic stem cell transplantation, which reported a similar sequential order of NKG2A and KIRs, independent of the HLA class I type of the patient.24 Finally, knockdown of HLA class I in MSCs by β2M-specific shRNA did not change this picture either and suggests that expression of HLA class I molecules in general is not significantly interfering with the initial formation of the NK cell receptor repertoire. In this context, it is of note that, in HLA class I–deficient patients, the overall frequency of KIR-expressing NK cells is not substantially different from healthy donors, with the exception of an increase in the NKG2A+KIR+ subset.25 It is unknown whether this phenotype is already present in these patients at birth or is established at later time points. Altogether, it appears likely that the sequential order of NK receptor expression is predominantly determined by intrinsic genetic factors. Unfortunately, the regulatory basis of this sequential acquisition process remains largely undetermined.
In summary, the MSC/HSPC coculture model presented here enables the study of NK cell differentiation in vitro in a human stem cell niche. In addition to representing a novel platform for analyzing basic questions of NK cell differentiation and NK cell repertoire adaptation, it enables efficient generation of large numbers of functional NK cells from HSPCs and, thus, could be of value in the rapidly emerging field of NK cell–based cellular therapies.26 Applications such as chimeric antigen receptor (CAR)-transfected NK cells represent promising new avenues in leukemia therapy.27 As shown here, 4 to 5 × 106 NK cells are routinely generated from 3000 HSPCs in MSC cocultures in 24-well plates. This protocol could be easily expanded to generate >1 × 109 NK cells from an average apheresis product. Moreover, the regulatory hurdles for generation of NK cells from CB-derived HSPCs in coculture with GMP-grade MSCs are much lower compared with the currently available protocols using murine-transformed and immortalized cell lines, such as OP9-DL1, EL08.1D2, or AFT024. Thus, transfection of HSPCs with CAR and subsequent MSC-supported NK cell differentiation could constitute a versatile platform to generate large numbers of CAR NK cells in GMP conditions. For example, this therapeutic avenue might be able to generate CAR NK cell products from leukemia patients who have otherwise strongly reduced or nonfunctional NK cell compartments.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Angela Manser (Institute for Transplantation Diagnostics and Cell Therapeutics) and Lutz Walter (German Primate Center, Göttingen, Germany) for input and critical comments.
This work was supported by the Deutsche Forschungsgemeinschaft (research grants UH91/7 and UH91/8) (M.U.) and the Research Commission of the Medical Faculty of Heinrich-Heine University (M.U.).
Authorship
Contribution: X.Z., J.B., M.H., S.W., and Ö.D. carried out the practical experiments; X.Z., G.K., R.M., and M.U. conceived the study, designed experiments and reviewed the data; M.U. wrote the manuscript; and all authors contributed to revising and editing the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Markus Uhrberg, Institute for Transplantation Diagnostics and Cell Therapeutics, Heinrich-Heine University, Medical Faculty, Moorenstr 5, D-40225 Düsseldorf, Germany; e-mail: markus.uhrberg@med.uni-duesseldorf.de.

