

Key Points
Plerixafor was administered to 6 red cell exchange transfused adults with SCD without complications.
Sufficient numbers of peripheral blood CD34+ cells could be collected by apheresis for potential use in gene therapy manufacturing.

Abstract
Novel therapies for sickle cell disease (SCD) based on genetically engineered autologous hematopoietic stem and progenitor cells (HSPCs) are critically dependent on a safe and effective strategy for cell procurement. We sought to assess the safety and efficacy of plerixafor when used in transfused patients with SCD for HSC mobilization. Six adult patients with SCD were recruited to receive a single dose of plerixafor, tested at lower than standard (180 µg/kg) and standard (240 µg/kg) doses, followed by CD34+ cell monitoring in peripheral blood and apheresis collection. The procedures were safe and well-tolerated. Mobilization was successful, with higher peripheral CD34+ cell counts in the standard vs the low-dose group. Among our 6 donors, we improved apheresis cell collection results by using a deep collection interface and starting apheresis within 4 hours after plerixafor administration. In the subjects who received a single standard dose of plerixafor and followed the optimized collection protocol, yields of up to 24.5 × 106 CD34+ cells/kg were achieved. Interestingly, the collected CD34+ cells were enriched in immunophenotypically defined long-term HSCs and early progenitors. Thus, we demonstrate that plerixafor can be employed safely in patients with SCD to obtain sufficient HSCs for potential use in gene therapy.
Introduction
Autologous cell-based therapies including lentiviral gene therapy and gene editing offer the possibility of cure for patients with sickle cell disease (SCD).1-6 Success of these therapies relies on safely obtaining autologous hematopoietic stem and progenitor cells (HSPCs), identified by expression of the CD34 marker, for genetic modification and transplantation. Availability of sufficient HSPCs is critical for adequate hematopoietic reconstitution and successful, timely engraftment of the engineered cells.
Procurement of HSPCs is uniquely challenging in patients with SCD. Bone marrow (BM) harvest under anesthesia carries a risk for SCD-related morbidity and may require repeated procedures to achieve a sufficient cell dose for manufacturing and infusion, particularly in adult subjects.7,8 In gene therapy trials for other diseases, HSCs are more abundantly obtained through peripheral blood (PB) collection after mobilization with granulocyte colony-stimulating factor (G-CSF). However, G-CSF is contraindicated in SCD9 because of severe adverse effects including vaso-occlusive crises, severe acute chest syndrome,10 massive splenomegaly, and death.11
Plerixafor is a mobilization agent that acts by reversibly inhibiting the binding of the chemokine stromal-derived factor 1 (SDF-1/CXCL12) to its receptor CXCR4, which is expressed on the surface of HSPCs.12 Plerixafor is safe and well-tolerated in healthy donors,13 and when combined with G-CSF in patients with lymphoma or multiple myeloma14-16 at a dose of 240 μg/kg. Plerixafor alone has been used as a salvage therapy in healthy allogeneic donors, with encouraging results.17 We tested whether plerixafor alone would be a safe mobilizing agent in SCD. After plerixafor mobilization, SCD donors underwent apheresis for collection of the mobilized CD34+ cells to test the safety and efficacy of the procedure and characterize the collected cells in this patient group.
Methods
Patients
Volunteer subjects were 18 to 40-year-old adults with SCD receiving regular exchange transfusions as part of existing medical care. Subjects with end-organ dysfunction, concurrent illnesses, or emergency room visits or hospitalizations for a SCD-related reason within 30 days were excluded. Patients taking hydroxyurea (HU) as part of their existing medical regimen were included and instructed to stop the HU 14 days before plerixafor administration.
Study design
A nonrandomized pilot safety and feasibility study (NCT02989701) was conducted under an Investigational New Drug (#131740) approved by the US Food and Drug Administration at Boston Children’s Hospital with Institutional Review Board approval. All participants gave written informed consent. Primary objectives were to describe the safety of mobilization with plerixafor and to assess the number of CD34+ cells collected in a single apheresis session. Within 7 days after their last exchange transfusion, to achieve a sickle hemoglobin (HbS) percentile of less than 30%, participants were admitted to the hospital (day −1) to receive plerixafor (day 0). The study included a dose escalation, with the first 3 subjects receiving 180 µg/kg plerixafor and then, in the absence of adverse events (AEs), the next 3 subjects receiving 240 μg/kg. Subjects began apheresis within 6 hours of plerixafor dosing (day 0) and were discharged day +1. They received intravenous hydration for the entire admission. When feasible, pre- and postplerixafor (preapheresis) bone marrow aspirates were performed. Bone marrow aspirates were obtained before and 3 to 4 hours after plerixafor administration.
Apheresis procedure
Apheresis was performed using a Cobe (Terumo) Spectra for the first 5 subjects and an Optia instrument for the sixth. Vascular access was either via peripheral intravenous or existing implanted venous access device (VAD). Anticoagulant citrate dextrose solution, solution A (1:10) was used as anticoagulant, and up to 5 blood volumes were targeted for collection.
Safety assessments
AE monitoring, vital signs, physical examination, and complete blood count with differential were performed during hospitalization, and outpatient on days +3, +7, and +14. AEs were assessed by telephone on day +2. AEs were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events v4.03.
Efficacy assessments
PB CD34+ cells/μL were measured by fluorescence-activated cell sorting analysis at baseline, on day −1, on day 0 preapheresis, every 2 hours during apheresis, and on day +1. CD34+ cell composition was determined by fluorescence-activated cell sorting, as previously reported.18 Collection products were processed and CD34+ cells selected using CliniMACS CD34 Reagent System per manufacturer (Miltenyi Biotec).
Immunophenotyping
Immunophenotype from healthy donor and SCD BM and of purified CD34+ products collected from mobilized PB samples was determined according to the gating strategy shown in Figure 3A, using conjugated antibodies from Biolegend (anti-human CD34, CD38, CD90, CD10, CD135, Lineage, CD15) and BD Bioscience (anti-human CD45RA and CD7). HSPC subpopulation proportions have been estimated by assuming a Dirichlet-multinomial distribution and using a maximum likelihood approach. Using parameter estimates and related standard errors, the comparison among different groups has been calculated by means of the Student t test under different variance hypothesis.
Results
Patient characteristics
Eight subjects were initially consented for the study. Two were excluded during the screening phase: 1 for personal scheduling conflicts and the other because of pregnancy. The trial participants (Table 1) were 6 adults, with an average age of 27 years (range, 19-38 years), and all were male with HbSS genotype. All subjects had been receiving routine exchange transfusions for from 3 to 23 years for the indication of primary stroke prophylaxis (n = 1) or secondary stroke prophylaxis (n = 5). None of the subjects were taking HU. Three subjects had previously undergone splenectomy. With the exception of 1 individual who had experienced a small number of vaso-occlusive pain events and episodes of acute chest syndrome, no other participants had experienced sickling complications in the 2 years before this study. Within 1 week before plerixafor administration, their postexchange transfusion percentage of HbS was below 30% (range, 5.5%-21.4%).
Baseline characteristics and cell collection yield of patients
| Subject | Age, y | Sex | No. VOE in last 2 y | No. ACS in last 2 y | History of splenectomy | History of CNS event | Resting CD34+* | Plerixafor dose, μg/kg | Peak CD34+, cells/μL | Plerixafor to apheresis time, h | CD34+ cells collected, ×106/kg | Ideal predicted CD34+ yield, × 106/kg† | CD34+ postselection recovery (purity), % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 30 | M | 2 | 2 | No | Yes | 1,2 | 180 | 36 | 5:42 | 0.069 | 3.1 | 81, (32) |
| 2 | 19 | M | 0 | 0 | No | Yes | 3,3 | 180 | 65 | 5:20 | 0.616 | 5.1 | 54, (84) |
| 3 | 26 | M | 0 | 0 | No | No | 2,4 | 180 | 31 | 5:35 | 1.2 | 2.6 | 20‡, (68) |
| 4 | 25 | M | 0 | 0 | Yes | Yes | 10,31 | 240 | 290 | 3:55 | 24.53 | 26.8 | 59, (99) |
| 5 | 38 | M | 0 | 0 | Yes | Yes | 2,2 | 240 | 27 | 3:54 | 2.94 | 2.2 | 41, (94) |
| 6 | 25 | M | 0 | 0 | Yes | Yes | 5,8 | 240 | 156 | 3:54 | 16.38 | 13.1 | 61, (99) |
| Subject | Age, y | Sex | No. VOE in last 2 y | No. ACS in last 2 y | History of splenectomy | History of CNS event | Resting CD34+* | Plerixafor dose, μg/kg | Peak CD34+, cells/μL | Plerixafor to apheresis time, h | CD34+ cells collected, ×106/kg | Ideal predicted CD34+ yield, × 106/kg† | CD34+ postselection recovery (purity), % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 30 | M | 2 | 2 | No | Yes | 1,2 | 180 | 36 | 5:42 | 0.069 | 3.1 | 81, (32) |
| 2 | 19 | M | 0 | 0 | No | Yes | 3,3 | 180 | 65 | 5:20 | 0.616 | 5.1 | 54, (84) |
| 3 | 26 | M | 0 | 0 | No | No | 2,4 | 180 | 31 | 5:35 | 1.2 | 2.6 | 20‡, (68) |
| 4 | 25 | M | 0 | 0 | Yes | Yes | 10,31 | 240 | 290 | 3:55 | 24.53 | 26.8 | 59, (99) |
| 5 | 38 | M | 0 | 0 | Yes | Yes | 2,2 | 240 | 27 | 3:54 | 2.94 | 2.2 | 41, (94) |
| 6 | 25 | M | 0 | 0 | Yes | Yes | 5,8 | 240 | 156 | 3:54 | 16.38 | 13.1 | 61, (99) |
ACS, acute chest syndrome; CNS, central nervous system; VOE, vaso-occlusive episode.
Multiple resting CD34+ counts were measured, all within 3 months before Plx. Lowest and highest are depicted here.
Predicted ideal collection yields were calculated; see also right half of Figure 1F.
Technical deviation from standard selection protocol.
Plerixafor mobilization was safe and allowed effective dose-dependent HSPC mobilization in SCD donors
The first 3 subjects received 180 µg/kg plerixafor subcutaneously preceded (for at least 6 hours) and followed (for ≥24 hours) by maintenance hydration with intravenous normal saline. No episodes of vaso-occlusive pain, acute chest syndrome, or other sickle cell-related events occurred up to the last follow-up visit at 14 days postmobilization. Therefore, the study proceeded as planned, with dose escalation to the 240 µg/kg dose in the subsequent 3 subjects, employing the same protocol. Similarly, no sickle cell-related events were observed after 240 µg/kg plerixafor dosing, up to the latest study follow-up. During the period of the study, subjects experienced the following AEs: grade 1 rash (n = 1), grade 1 cough (n = 1), grade 2 headache (n = 1), grade 3 anemia (before plerixafor administration; n = 1), and grade 3 fever (before plerixafor administration; n = 1). No AE associated with plerixafor, including diarrhea, persistent hyperleukocytosis, or thrombocytopenia, was observed in any subjects.
After plerixafor administration, the total white blood cell (WBC) count increased 2.6-fold over the baseline values (range, 2.2- to 2.9-fold) in the 180 µg/kg group. In the 240 µg/kg group, there was a similar 2.4-fold increase of the WBC counts over baseline values (range, 2.1- to 2.9-fold). In all patients, WBC counts progressively returned to near baseline levels by day +1 (Figure 1A). A parallel increase in the absolute neutrophil and lymphocyte counts was observed in PB of the donors, as depicted in Figure 1B-C.
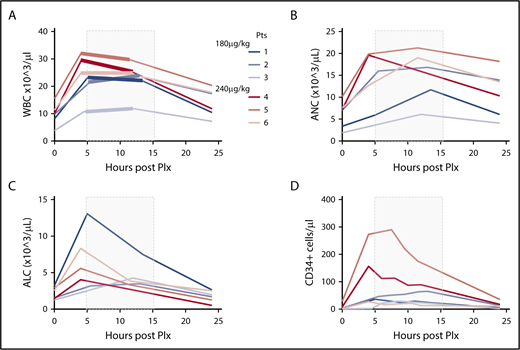
PB cell characteristics after mobilization. (A-D) PB cell counts for WBC (A), absolute neutrophil count (ANC) (B), absolute lymphocyte count (ALC) (C), and CD34+ cells (D) were obtained immediately before mobilization and after plerixafor (Plx) administration during apheresis at the indicated time intervals. The shaded gray rectangle represents the period of apheresis collection. The thickened portions of the colored lines in panel A indicate precise beginning and end times for apheresis in each subject.
PB cell characteristics after mobilization. (A-D) PB cell counts for WBC (A), absolute neutrophil count (ANC) (B), absolute lymphocyte count (ALC) (C), and CD34+ cells (D) were obtained immediately before mobilization and after plerixafor (Plx) administration during apheresis at the indicated time intervals. The shaded gray rectangle represents the period of apheresis collection. The thickened portions of the colored lines in panel A indicate precise beginning and end times for apheresis in each subject.
PB CD34+ counts preceding mobilization varied substantially among individuals in a range of 1 to 28 cells/µL (Table 1). In the 180 µg/kg group, starting 4 hours after plerixafor, an increase of PB CD34+ cell count over baseline values was observed, stable during the first 12 hours after plerixafor administration, with raw values reaching an average peak of 44 CD34+ cells/µL (range, 31-65 cells/µL). In the 240 µg/kg group, starting 4 hours and up to 12 hours after plerixafor administration, PB CD34+ cell counts over baseline values were observed, with raw values reaching an average peak of 158 CD34+ cells/µL (range, 27-290 cells/µL), with an average 14.5-fold increase over baseline values (range, 10.4- to 19.5-fold; Figure 1D). In all patients, CD34 cell counts progressively returned to near baseline levels by day +1 (Figure 1D). A plerixafor dose effect was identified with statistically significantly higher PB CD34+ cell counts with 240 µg/kg than with 180 µg/kg (P = .0068; Figure 2A).
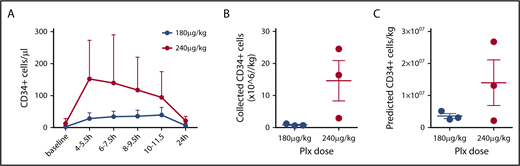
Mobilization and collection by apheresis. (A) PB CD34+ values were stratified by dose. A significant effect of Plx dose on PB CD34+ cell counts was observed by 2-way analysis of variance (P = .0068). Averages are shown with bars representing range. Total collected CD34+ cells (B) and predicted ideal collection yield based on an average PB CD34+ cell count during apheresis (C) for each dose are shown. Average ± standard error of the mean; (n = 3 at 180 µg/kg, n = 3 at 240 µg/kg).
Mobilization and collection by apheresis. (A) PB CD34+ values were stratified by dose. A significant effect of Plx dose on PB CD34+ cell counts was observed by 2-way analysis of variance (P = .0068). Averages are shown with bars representing range. Total collected CD34+ cells (B) and predicted ideal collection yield based on an average PB CD34+ cell count during apheresis (C) for each dose are shown. Average ± standard error of the mean; (n = 3 at 180 µg/kg, n = 3 at 240 µg/kg).
An optimized apheresis procedure allowed effective HSPC collection in SCD mobilized donors
Apheresis was performed on mobilized subjects in the 180 µg/kg cohort within 6 hours after plerixafor administration. After observing early mobilization in these patients, we elected to start apheresis at 4 hours after plerixafor administration in the 240 µg/kg cohort. We performed apheresis using the COBE Spectra instrument for the first 5 donors, together with the manufacturer’s WBC collection kit. The sixth subject was collected using an Optia instrument, although no significant change was found in efficiency of collection.
For the first 2 subjects, we chose a standard HSPC collection interface of 3%, as recommended by the manufacturer and used for routine HSPC collections in our center. Collection efficiencies in healthy donors using these settings rarely fall below 40%. Initial product CD34+ collection yields in these 2 subjects were far lower than expected, with fewer than 1 × 106 cells/kg (Table 1), despite both subjects having substantial PB mobilization of CD34+ cells. There were no mechanical interruptions during the procedure, and venous access was maintained. Between 2.5 to 3.5 blood volumes were processed (Table 2). Given the poor yield of CD34+ cells in the apheresis product, we altered our collection strategy based on reports of collections in patients with β thalassemia19-22 or microcytosis23 that suggested altered sedimentation of HSCs. In these patient populations, interface parameters of 5% to 10% were reported to improve recovery of CD34+ cells. Thus, a deeper red cell collection between 5% and 10% interface was used in the subsequent 4 subjects with significantly higher CD34+ yields (Table 2). Previous reports of predictors of collection efficiency postulated that the lymphocyte to neutrophil ratios would predict poor collectors.21 We calculated these ratios (Table 2), but did not find sustained values greater than 1. A single value for patient 1 was found to be 2.2 only at initiation of apheresis, and fell below 1 at all other times. We also tested whether collection efficiency was affected by percentage HbS remaining after the red cell exchange procedure (Table 2); we found no direct correlation. Taken together, we could not find a unifying parameter that could predict our low efficiencies using standard collection methods. Strikingly, the modifications introduced in the 4 subjects collected after the interface adjustment resulted in a markedly improved collection yield, which in 2 instances surpassed our ideal predicted collection efficiency of 40% (Table 1; Figure 2C).
Collection parameters
| Subject | Red cells in product, %Hct | L/N ratio* | %HbS before collection† | Blood volume processed, L |
|---|---|---|---|---|
| 1 | 3.7 | 2.2 | 5.5 | 3.0 |
| 2 | 5.6 | 0.2 | 13.3 | 2.5 |
| 3 | 17 | 0.5 | 9.9 | 3.5 |
| 4 | 10.5 | 0.3 | 21.4 | 4.0 |
| 5 | 16.4 | 0.7 | 6.9 | 3.2 |
| 6 | 11.7 | 0.2 | 9.2 | 4.0 |
| Subject | Red cells in product, %Hct | L/N ratio* | %HbS before collection† | Blood volume processed, L |
|---|---|---|---|---|
| 1 | 3.7 | 2.2 | 5.5 | 3.0 |
| 2 | 5.6 | 0.2 | 13.3 | 2.5 |
| 3 | 17 | 0.5 | 9.9 | 3.5 |
| 4 | 10.5 | 0.3 | 21.4 | 4.0 |
| 5 | 16.4 | 0.7 | 6.9 | 3.2 |
| 6 | 11.7 | 0.2 | 9.2 | 4.0 |
Hct, hematocrit.
Based on Constantinou et al.21
HbS was measured at the end of red cell exchange within 1 week of collection.
Overall predicted cell yield of the procedure can be sufficient for gene therapy dosing
Volume of blood processed and stable blood flow during apheresis are important parameters for efficient HSPC collections. Collection processing volumes were targeted for at least 5 blood volumes and up to 8 hours, to allow for patient safety in this pilot trial. We used existing venous access devices or peripheral vein accesses, which limited our flow to a maximum of 40 to 50/mL per minute. In subjects 1 and 2, flow was intermittently impaired because of VAD dynamics necessitating peripheral intravenous access after the start of the procedure. Given the technical limitations related to access and differing collection interfaces among the 6 subjects, we calculated the predicted ideal collection yields estimated using a formula based on each subject’s average PB CD34+ count during apheresis, ideal blood volume of 5.2 L (70 kg male), and ideal processed blood volume of 24 L.24 This calculation predicts that 2 of 3 subjects in the 240 µg/kg group would generate more than 10 × 106 CD34/kg (a standard target dose for cell manufacturing and back up in gene therapy) in a single apheresis session, whereas the other subjects would require repeated collections to meet this target (Figure 2B; Table 1). All apheresis products underwent CD34+ selection using Clinimacs-based procedure (Miltenyi Biotec),25 and recovery was assessed. Recovery varied widely (Table 1); however, for the 3 products collected using an altered collection protocol that yielded a product with an average hematocrit of about 12% (subjects 4-6, Table 2), CD34+ recovery averaged 54%, which is similar to values we and others obtain for non-sickle cell products.25
The plerixafor-mobilized apheresis product is enriched in HSCs
An ideal gene therapy product would contain a relatively high percentage of lineage-unspecified progenitors/bona fide hematopoietic stem cells to guarantee effective long-term, multilineage repopulation of recipient hematopoiesis. To assess whether plerixafor use could yield an apheresis product enriched in HSCs, we characterized our samples by immunophenotyping. As 3 of our subjects underwent BM aspiration before (in 2 of them) and after (in 3) plerixafor administration, we characterized by the same approach for these pre- and postplerixafor BM samples to evaluate the effect of plerixafor. For this purpose, we employed a multicolor flow cytometry strategy allowing the identification of multiple subpopulations within the CD34+ cell subset, corresponding to cells transitioning from the HSC stage to lineage-committed progenitors, using a combination of surface markers described in Doulatov et al18 (Figure 3A). Comparison with healthy donor BM- and mobilized PB-derived CD34+ purified cells was performed. We observed no clear differences in the proportion of HSPC subfractions contained in the SCD preplerixafor BM-derived CD34+ samples compared with age- and sex-matched healthy donors (P > .05; Figure 3B; supplemental Table 1). In the subjects with both pre- and postplerixafor BM samples, there was no marked change in HSC frequency after plerixafor administration (P > .05; Figure 3B). We then sought to characterize cell populations in CD34-purified apheresis products derived from healthy donors mobilized with G-CSF, and from patients with SCD mobilized with plerixafor. CD34+ cells collected from plerixafor-mobilized patients with SCD were enriched in HSPCs compared with healthy donor G-CSF-mobilized apheresis products (P = .0012; Figure 3B), with 3.7% (HSC) in healthy donor vs 9.7% in SCD products. These findings indicate that plerixafor allows the collection of ideal apheresis products for gene therapy applications in patients with SCD.
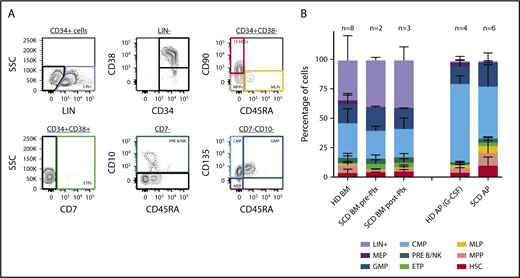
Immunophenotype ofproducts. We employed immunophenotyping according to the gating strategy shown in panel A to characterize the cell products collected from healthy donor (HD) bone marrow (BM; n = 8), HD mobilized apheresis product (AP) obtained after G-CSF stimulation (n = 4), SCD BM pre-Plx (n = 2) and post-Plx (n = 3), and SCD AP obtained after Plx administration (n = 6). Bone marrow products are unselected and apheresis products are CD34+ selected (B).
Immunophenotype ofproducts. We employed immunophenotyping according to the gating strategy shown in panel A to characterize the cell products collected from healthy donor (HD) bone marrow (BM; n = 8), HD mobilized apheresis product (AP) obtained after G-CSF stimulation (n = 4), SCD BM pre-Plx (n = 2) and post-Plx (n = 3), and SCD AP obtained after Plx administration (n = 6). Bone marrow products are unselected and apheresis products are CD34+ selected (B).
Discussion
Our study in a small number of patients demonstrates that plerixafor is safe and well-tolerated in exchange-transfused patients with SCD and allows collection of HSPC cells enriched in long-term HSCs in adequate quantities for gene therapy protocols. The subjects in our study experienced no vaso-occlusive pain or other sickle cell-related AEs after plerixafor administration. We took several precautions to minimize the possibility of sickling complications, including a premobilization exchange transfusion within a short time frame of the procedure, as well as an inpatient stay for extensive pre- and posthydration, and these measures may be advisable for patients with sickle cell in future clinical trials.
Mobilization as measured by peripheral CD34+ count was successful after plerixafor alone, and we noted a wide range of peak CD34+ concentrations, as has been documented in other patient populations undergoing peripheral mobilization. Although we first tested a lower dose in our trial, the standard dose of 240 µg/kg caused no AEs and resulted in higher average peripheral CD34+ counts, which suggests this dose may be preferable for use in gene therapy. In future studies, characterization of resting CD34+ count, WBC differential, and other cellular components of PB in larger cohorts of patients with SCD could provide prognostic markers to identify good mobilizers, and a higher plerixafor dose26 could be tested in this patient population for use in poor mobilizers.
Importantly, in our study, collection procedures of the mobilized HSPCs were optimized in several ways on the basis of the characteristics of patients with SCD and products. First, based on previous thalassemia experience, we adjusted the apheresis protocol to target a deeper collection interface, which resulted in higher yields and preserved the recovery and purity achieved. Importantly, the increased number of red cells in the apheresis product resulting from this altered collection strategy does not negatively affect the CD34+ selection process or the quality of the product. We estimate that an additional 50 mL of red cells are lost during collection at a deeper interface, which would require monitoring in patients undergoing multiday apheresis sessions. Thus, this is a feasible collection strategy for broad application in clinical trials of autologous genetic therapies in SCD. The variables affecting less efficient apheresis collections in patients with SCD remain unknown, and future studies may help identify subsets of patients that may not need collections at deeper interfaces. Second, our study highlighted the critical importance of optimal vascular access, especially for patients with relatively low mobilization who may require longer collections and larger product volumes. Vascular access was problematic in some early subjects in our study, in which we used combinations of existing VADs and peripheral access, rather than an apheresis-grade catheter for collection. A dedicated VAD for apheresis in all subjects would allow for higher volumes of blood processed in each session, which was a major limitation in our study. Third, based on the observation that HSPCs were already mobilized at 4 hours after plerixafor, earlier than previously described, we implemented an earlier apheresis start time 4 hours after plerixafor administration.
By optimizing the collection process as described, at the standard plerixafor dose, a single apheresis session yielded 2.94 to 24.5 × 106 CD34+ cells/kg, a range that would allow collection of a sufficient total number of HSPCs for most current gene therapy trials via single (in 2 of 3 cases) or sequential (in 1 case) collections. Indeed, using this protocol, we have successfully collected and manufactured 3 products for infusion in an ongoing gene therapy trial targeting BCL11A in patients with SCD (E.B.E., A.B., and D.A.W., 31 August 2018, data not shown; NCT03282656).
Our study included a comprehensive characterization of cell progenitors by immunophenotyping of the collected product, as well as of BM samples from the enrolled subjects before and after plerixafor mobilization. We did not find a difference in HSC and subpopulation proportions in the BM after plerixafor administration in the 2 patients with SCD examined, which would not support its use to improve CD34+ cell recovery in BM harvests. Furthermore, our cell population analyses show similar overall proportions of HSCs and precursors between healthy patients and patients with SCD. Interestingly, the analysis of our apheresis collection products revealed that cells collected from subjects with SCD after plerixafor mobilization are highly enriched in immunophenotypically defined HSCs (and multipotent progenitors), as compared with BM samples and G-CSF apheresis products collected from healthy donors. These products thus represent an ideal target cell population for use in gene therapy applications potentially associated with a greater probability for long-term engraftment and multilineage differentiation of the transduced cells as compared with more traditional products.
During the publication of this study, 3 additional groups reported the use of plerixafor in patients with SCD. Boulad et al reported interim results from a dose escalation study in which 15 patients with SCD were treated with a plerixafor dose of 80, 160, or 240 µg/kg.27 No apheresis collection was performed. There were 2 episodes of vaso-occlusive events (pain) associated with the plerixafor administration in this study. A second group reported 3 patients given plerixafor at 240 µg/kg and then collected by apheresis.28 Two of the 3 patients experienced grade 3 AEs associated with either the mobilization or collection in this study; however, all 3 had CD34 yields of greater than 5 × 106/kg. In a third publication, Lagresle-Peyrou et al reported a study of 3 adults with SCD who were exchange-transfused for 2 months before collection, were treated with standard dose plerixafor, and then underwent apheresis.29 No AEs were reported with the mobilization and collection in this study. Similar to our study, Lagresle-Peyrou et al29 noted early mobilization, whereas Boulad et al27 reported initial measurements of CD34+ cells only 6 to 12 hours after plerixafor.
These additional studies highlight the importance of considering the effect of HU on HSC collection. Because many patients with SCD considering gene therapy will be receiving a HU regimen at baseline, the question of whether and how this medication affects peripheral stem cell mobilization and collection is important. Data from a small study in patients with SCD showed an increase in peripherally circulating CD34+ cells 14 days after withdrawal of HU.30 Patients with thalassemia who receive HU and who underwent plerixafor mobilization19 did not mobilize successfully unless they had a washout period of at least 2 weeks before mobilization and collection, suggesting that the myelosuppressive effect of HU could negatively affect HSPC procurement. In our small pilot trial reported here, the subjects were not taking HU at the time of mobilization for 2 reasons. First, we opted to only include patients already on chronic exchange transfusion regimens, and in practice, patients receiving chronic transfusions are far less likely to also be prescribed HU. In addition, for any subjects who were taking HU, we designed our study to mandate cessation of the medication 14 days before plerixafor administration. Similar to our study, the 3 subjects in Legresle-Peyrou et al29 were not taking HU and were given at least 3 months of transfusions before mobilization. In contrast, two-thirds of the patients in Boulad et al’s study27 were receiving HU, and only 1 of 15 patients received transfusions before mobilization. Although there was no statistically significant difference in WBC count or CD34+ count after plerixafor between patients taking or not taking HU in their study, it is possible that the lower average mobilization achieved in that study is, in part, attributable to myelosuppression from HU. As supported by these studies and suggested by others,31 future clinical trials for autologous genetic therapies in sickle cell disease should consider a preparative regimen of transfusions and HU cessation.
In conclusion, the reassuring safety profile demonstrated in this study and in the other recently published experiences27-29 suggests that collection of HSPC by apheresis may be both safer and more tolerable than multiple bone marrow harvests. This study supports standard-dose plerixafor mobilization, followed by apheresis, using an optimized protocol as a safe and effective approach for collection of autologous HSPCs for use in gene therapy and transplantation in patients with SCD.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Colleen Dansereau, Brenda MacKinnon, Stephanie Patriarca, and Felicia Ciuculescu for clinical research support; Maureen Achebe for patient referral; Erdyni Tsitsikov for technical support; BCH Therapeutic Apheresis Unit and Clinical and Translational Study Unit teams for clinical care; Danilo Pellin for statistical support on immunophenotype studies; Christian Brendel, Dan Bauer, and the Data and Safety Monitoring Board members (Philip Breitfield, John DiPersio, Orah Platt, and Shalini Shenoy) for fruitful discussion of the data.
This work was supported by research funding from Bluebird bio, and by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL137848) (D.A.W.) and the Boston Children’s Hospital Translational Research Program Pilot (E.B.E. and D.A.W.)
Authorship
Contribution: E.B.E., J.P.M., and A.B. designed the research, conducted the trial, and wrote the manuscript; H.D., C.B., H.T.-N., F.J.P., M.A., S.N., M.M.H., and L.B. performed research; W.B.L. contributed to statistical design and data analysis; M.A. and D.A.W. supported research; and all authors revised the manuscript.
Conflict-of-interest disclosure: D.A.W. has licensed technology related to gene therapy of hemoglobinopathies to Bluebird bio and has received funding via a sponsored research agreement as part of this license agreement. A.B. and E.B.E. have received funding from Bluebird bio via a sponsored research agreement to conduct the described clinical trial. H.T.-N.’s spouse is employed by Bluebird bio. M.A. and F.J.P. are employees of Bluebird bio. The remaining authors declare no competing financial interests.
Correspondence: Alessandra Biffi, Dana-Farber/Boston Children's Cancer and Blood Disorders Center, 450 Brookline Ave, Boston MA 02215; e-mail: alessandra.biffi@childrens.harvard.edu.

