

Key Points
Constitutional mismatch repair deficiency syndrome should be considered in children with acute leukemia and characteristic skin lesions.
The high mutation burden of CMMRD-related cancers contributes to treatment resistance, necessitating individualized treatment strategies.
Introduction
B-cell precursor acute lymphoblastic leukemia (B-ALL) is the most common childhood cancer.1 Although most childhood B-ALL is sporadic, a subset occurs in children with preexisting conditions that predispose to leukemogenesis.2 Cancer predisposition syndromes associated with chromosomal instability with syndromic features, such as Bloom syndrome, Fanconi anemia, and Nijmegen breakage syndrome, are often recognized prior to the development of cancer. By contrast, Li-Fraumeni syndrome and congenital mismatch repair deficiency (CMMRD) syndrome can be more challenging to diagnose without family history, and childhood cancer may be the presenting finding.
B-ALL that develops in the context of a cancer-predisposition syndrome presents unique challenges. Chemotherapy resistance, impaired chemotherapy tolerance, and magnified susceptibility to acute/late treatment effects require individualized treatment approaches.3 Issues surrounding testing/counseling of potentially affected family members, as well as psychological impacts of diagnosis, cannot be overlooked. Here, we present the clinical course of a young patient with CMMRD-related B-ALL that highlights these complexities.
Case description
At 7 years of age, the patient developed easy bruising; after a complete blood count identified thrombocytopenia and leukocytosis, a bone marrow evaluation was performed, confirming the diagnosis of B-ALL, with 93.5% blasts by manual differential. Multicolor flow cytometry identified an abnormal precursor B-cell population expressing CD19, CD20, CD24, CD10, CD9, CD34, CD38, CD15, and HLA-DR. Cytogenetic evaluation of leukemic blasts demonstrated deletion within chromosome 7p, confirmed by microarray analysis to be between bands p14.1 and p11.2, a region containing the IKZF1 gene. This high-risk genetic finding, along with residual leukemia persistence after induction therapy, prompted intensification to high-risk therapy.
Physical examination was notable for multiple hyper- and hypopigmented macules scattered on his trunk and extremities, without axillary freckling. Previously, a genetic evaluation to assess for neurofibromatosis type 1 (NF1) and Noonan syndrome was unrevealing. He did not develop additional criteria for NF1, and there were no affected family members. The development of an early malignancy, along with conspicuous skin findings, prompted investigations for mismatch repair defects. Mutational analysis of MLH1, MSH2, MSH6, and PMS2 revealed biallelic mutations in the PMS2 gene with the homozygous pathogenic variant c.1831dupA, diagnostic of CMMRD. Both parents were found to be heterozygous with the same mutation, consistent with hereditary nonpolyposis colorectal cancer syndrome or Lynch syndrome. Appropriate screening was initiated for the patient and parents.
During maintenance chemotherapy, the patient experienced relapse with the same immunophenotype as at diagnosis. Conventional karyotyping and chromosomal microarray were unchanged, without additional abnormalities. Reinduction attempts with conventional chemotherapy failed to achieve a second remission. Despite sequential treatment with the CD19-directed bispecific T-cell–engaging agent blinatumomab and CD22-directed inotuzumab ozogamicin, the leukemia progressed. Ultimately, the patient died of complications of progressive disease.
Methods
DNA was isolated from bone marrow samples collected at presentation, remission, and first relapse. DNA was quantified using a Qubit dsDNA HS Assay Kit and Qubit fluorometer (both from Thermo Fisher Scientific). Five-hundred nanograms of genomic DNA from both tumor and normal tissues was submitted to the Center for Applied Genomics at the Hospital for Sick Children for whole-exome sequencing (WES), alignment to the reference genome, and variant calling. Agilent’s SureSelect All Exon V5 kit was used for enrichment, and paired-end sequencing was done on an Illumina HiSeq 2500. The software bcl2fastq2 v2.17 was used to generate raw fastq files. Alignment to the hg19 reference genome was done using BW-MEM 0.7.12, followed by Picard Tools 1.133 to mark duplicates and GATK 3.4-46 IndelRealigner for local matched normal samples to call somatic single nucleotide variants (SNVs), and followed by annotation using ANNOVAR (version February 2015). The tumor mutation burden (TMB) (mutations per megabase) from WES was calculated by counting the total number of somatic SNVs and dividing by the total number of callable bases in megabases. deconstructSigs4 was used to determine COSMIC signatures5 in the mutation spectrum within a trinucleotide context for each sample. All analyses were done in R version 3.4.3 using the high-performance computing cluster at the Hospital for Sick Children.
Results and discussion
This clinical scenario highlights challenging aspects in diagnosis and management of B-ALL in the context of CMMRD. CMMRD results from germline biallelic mutations in 1 of the mismatch repair genes (MLH1, MSH2, MSH6, or PMS2). Potential malignancies include central nervous system tumors, hematologic malignancies, and gastrointestinal cancers; in general, the initial cancer diagnosis is established in the first decade of life (median, 7.5 years),6 and few patients survive into adulthood. Hematologic malignancies are common and typically are lymphomas of T-cell origin; however, B-cell malignancies are being increasingly observed.7
Carcinogenesis in CMMRD is mediated by impaired correction of mutations and microsatellite instability resulting from errors during DNA replication.8 As such, in contrast to other DNA repair/instability disorders, in CMMRD there are currently no recommendations to avoid particular chemotherapeutic agents or radiation over concerns for excess toxicity.9 However, malignancies associated with CMMRD are known to be relatively resistant to certain chemotherapy agents, notably including antimetabolites, such as mercaptopurine.10 Of note, our patient did not develop unexpected or severe chemotherapy-related toxicities connected to CMMRD.
In addition to chemotherapy resistance, CMMRD-related malignancies are associated with relatively high TMB.11-13 Indeed, genomic analysis of WES data from the diagnostic leukemia specimen of our patient revealed 9.4 mutations per megabase, which represents a remarkably high TMB, particularly in the context of pediatric B-ALL. Although conventional cytogenetic and microarray evaluation did not identify new genetic lesions at the time of relapse, subsequent genomic analysis showed that the relapse sample had twice the TMB of the diagnostic sample (19.6 mutations per megabase), indicating rapid accumulation of additional mutations in residual leukemic blasts or the expansion of an ancestral clone with a higher TMB conferring treatment resistance (Figure 1). Hypermutation, typically defined as >10 mutations per megabase, is relatively rare in pediatric tumors in general, particularly in hematologic malignancies, which are known for very low TMB.13 Similarly, an elevated number of SNVs were detected in the relapse sample compared with the primary sample (953 vs 457, respectively); of these, 98 SNVs were common to both time points (Figure 1).
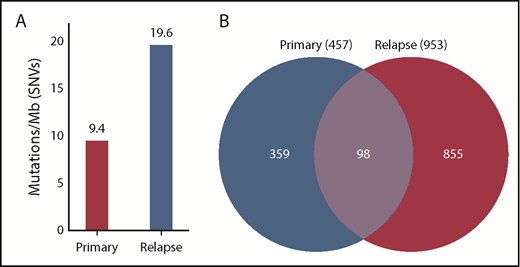
Mutation burden at diagnosis and relapse. DNA was extracted from primary, remission, and relapse samples and subjected to WES. The remission sample was used as a baseline control to exclude germline variants. (A) SNVs were quantified, and TMB was calculated, showing that the primary sample contained 9.4 mutations per megabase of DNA, whereas the relapse sample had 19.6 mutations per megabase. (B) A total of 457 SNVs was detected in the primary sample, and 953 SNVs were detected in the relapse sample. Of these, 98 SNVs were common to the primary and the relapse samples.
Mutation burden at diagnosis and relapse. DNA was extracted from primary, remission, and relapse samples and subjected to WES. The remission sample was used as a baseline control to exclude germline variants. (A) SNVs were quantified, and TMB was calculated, showing that the primary sample contained 9.4 mutations per megabase of DNA, whereas the relapse sample had 19.6 mutations per megabase. (B) A total of 457 SNVs was detected in the primary sample, and 953 SNVs were detected in the relapse sample. Of these, 98 SNVs were common to the primary and the relapse samples.
The hypermutant phenotype can result in loss of important antigens targeted by immunotherapies, such as blinatumomab, inotuzumab ozogamicin, and chimeric antigen receptor T cells. Indeed, after exposure to antigen-directed agents, the patient’s leukemia cells rapidly progressed and exhibited antigen loss with development of a CD19− subpopulation after blinatumomab (Figure 2) or decreased antigen density of CD22 after inotuzumab. This suggests multiple subclones with varying antigenic expression or a rapidly evolving tumor acquiring methods of treatment escape. Notably, in general, CD19− relapse occurs in only a minority of blinatumomab nonresponders.14
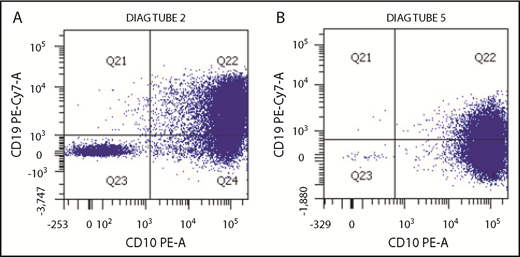
Change in leukemic blast expression of CD19 after selective pressure of blinatumomab. (A) Multicolor flow cytometric analysis of leukemia blasts after conventional chemotherapy exposure demonstrates predominantly normal CD19 expression. (B) After exposure to the CD19-directed immunotherapeutic agent blinatumomab, there is a large CD19− blast population. This correlates with the progression of disease seen during administration of blinatumomab, possibly as a result of hypermutation of the malignant clone.
Change in leukemic blast expression of CD19 after selective pressure of blinatumomab. (A) Multicolor flow cytometric analysis of leukemia blasts after conventional chemotherapy exposure demonstrates predominantly normal CD19 expression. (B) After exposure to the CD19-directed immunotherapeutic agent blinatumomab, there is a large CD19− blast population. This correlates with the progression of disease seen during administration of blinatumomab, possibly as a result of hypermutation of the malignant clone.
Hypermutant tumors may express tumor-specific neoantigens, which can potentially be recognized by immune effector cells and targeted for killing. This, along with the known phenomenon of upregulation of immune checkpoint molecules, such as PD-1 and CTLA-4,15 makes immune checkpoint inhibitors attractive therapeutic options in these patients.16,17 In addition, it is tempting to speculate that allogeneic hematopoietic cell transplantation and the attendant polyclonal alloreactive graft-versus-leukemia effect may have facilitated disease control, balanced against risks of treatment-related mortality and late effects, such as second malignancies.18 Because tolerance of intensive chemotherapy and radiation is not impaired in CMMRD, a fully myeloablative regimen with rapid taper of immunosuppression would have been possible, optimizing the likelihood of successful transplantation. Immunologic therapies such as these, with a broad range of target antigens, may be less susceptible to resistance than single antigen-targeted therapies, such as those used in this patient.
In summary, B-ALL in the context of CMMRD presents distinct therapeutic challenges related to high TMB and acquisition of additional mutations under selective treatment pressure. Early recognition of CMMRD and individualization of therapy are critical for successful management of these patients. Finally, novel approaches are needed to optimize care in this high-risk setting and improve survival.
Authorship
Contribution: B.O. participated in the care of the patient, conceived the idea for publication, and wrote the manuscript; N.G., C.M., J.N., and M.C. participated in the clinical care of the patient and contributed to the manuscript; M.E., S.S., and V.J.F. performed the genomic analysis of tumor specimens and contributed to the manuscript; and U.T. guided the direction of the report, oversaw the genomic analysis, and contributed to the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Pediatric Cancer and Blood Disorders Institute, Benjamin Oshrine, Johns Hopkins All Children’s Hospital, 601 5th St South, Suite 302, St. Petersburg, FL 33701; e-mail: boshrin1@jhmi.edu.

