

Key Points
Absence of C3 does not prevent classical pathway–mediated hemolysis.
Absence of C3 abolishes alternative pathway–mediated hemolysis.

Abstract
Complement component 3 (C3) is emerging as a potential therapeutic target. We studied complement-mediated hemolysis using normal and C3-depleted human sera, wild-type (WT) and C3-deficient rat sera, and WT and C3 knockout rat models. In all of the in vitro and in vivo experiments, we found that the loss of C3 did not prevent classical pathway–mediated hemolysis, but it did almost abolish alternative pathway–mediated hemolysis. Experiments using preassembled classical pathway C3 convertases confirmed that C4b2a directly activated complement component 5 (C5), leading to membrane attack complex formation and hemolysis. Our results suggest that targeting C3 should effectively inhibit hemolysis and tissue damage mediated by the alternative pathway of complement activation, but this approach might have limited efficacy in treating classical pathway–mediated pathological conditions.
Introduction
Complement, a key part of the innate immune system, is composed of multiple components and needs to be activated to function. Complement can be activated through 3 major activation pathways: classical, alternative, and lectin. These activation pathways play different roles in the pathogenesis of different diseases. Complement activation is a critical means of host defense against infection and the clearance of immune complexes.1 However, excessive complement activation causes tissue damage such as hemolysis, which is seen in diseases such as paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome, and cold agglutinin disease (CAD).2 Consequently, efforts are underway to develop inhibitors that target different complement components as novel therapeutic agents. For example, eculizumab, a monoclonal antibody specific for complement component 5 (C5) has been approved for clinical use and was shown to effectively reduce complement-mediated hemolysis in patients with PNH, atypical hemolytic uremic syndrome, or CAD.3
Complement component 3 (C3) is the central component of all 3 major complement activation pathways required for both complement-mediated opsonization and membrane attack complex (MAC) formation. C3 has generated considerable interest as another promising target for the treatment of diseases in which complement is an integral pathogenic mechanism, including diseases associated with complement-mediated hemolysis.4 This concept has been supported by studies including those using C3 knockout (KO) mice or C3 inhibitors in mice, in which complement-mediated hemolysis (both extravascular and intravascular) was shown to be significantly reduced in various models in the absence or inhibition of mouse C3.5-7 However, the hemolytic activity of mouse complement is 200- to 300-fold lower than that of human complement,8 and therefore the mitigation of complement-mediated hemolysis observed in C3 KO or C3-inhibited mice might not represent the actual situation in humans. Because the hemolytic activity of rat complement is comparable to that of human complement8 and because we recently developed a C3 KO rat,9 we investigated complement-mediated hemolysis using wild-type (WT) and C3 KO rats as well as normal and C3-depleted (C3-Dpl) human sera to clarify the rationale for the development of C3-targeted therapeutics.
Materials and methods
C3-deficient rats and sera
C3 KO rats were developed and characterized as described before.9 Age- and sex-matched WT and C3 KO rat littermates were used in all experiments. All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee of Cleveland Clinic. Pooled normal human sera (NHS) and C3-Dpl human sera were purchased from Complement Technology Inc. (Tyler, TX). No C3 protein was detectable by western blot in the C3-Dpl human sera.
In vitro classical pathway complement–mediated hemolytic assay
Sheep red blood cells (RBCs) (Hemostat Laboratories, Dixon, CA) were first sensitized with rabbit anti-sheep RBC serum (MP Biomedicals, Santa Ana, CA). Approximately 5 × 106 sensitized sheep RBCs (EshA) were incubated with either NHS or C3-Dpl sera (0.5%-100%) in gelatin veronal buffer with Mg++ and Ca++ (GVB++; 10 mM barbital, 145 mM NaCl, 0.5 mM MgCl2, 0.15 mM CaCl2, gelatin 0.1%, pH 7.2 ± 0.15; Boston BioProducts, Ashland, MA) at 37°C. Then, 5 mM EDTA was added to the buffer to inhibit the complement activity in negative controls. Hemolysis mediated by the WT and C3 KO rat sera was tested following the same procedures.
After incubation (20 minutes for low sera concentrations [0.5%-10%] and 5 minutes for high sera concentrations [20%-100%]), EshA cells were centrifuged and the supernatants were collected for optical density (OD) measurement at 414 nm (OD414). The following equation was used to calculate the percentage of hemolysis: hemolysis (%) = [(A − B)/(C − B)] × 100%. A is the OD reading of the sample with sera, B is the OD reading of the sample with sera and EDTA, and C is the OD reading of maximum hemolysis induced by H2O.
In some experiments, freshly prepared rat RBCs were sensitized with a rabbit anti-rat RBC anti-serum (1:2) (Cedarlane Laboratories, Burlington, NC) and incubated with different concentrations of WT and C3 KO rat sera (5%-100%) in GVB++ buffer at 37°C for 60 minutes. Complement-mediated hemolysis was calculated using the same method.
In vitro alternative pathway complement–mediated hemolytic assay
Rabbit RBCs (Erabb) (Hemostat Laboratories, Dixon, CA) were incubated with GVB-Mg-EGTA (5 mM barbital, 145 mM NaCl, 0.5 mM MgCl2, 10 mM EGTA, and 0.1% gelatin, pH 7.2 ± 0.15; Boston BioProducts) with 5% to 100% of either NHS or C3-Dpl human sera and WT or C3 KO rat sera at 37°C for 20 minutes. The percentage of hemolysis was calculated using the equation described in “in vitro classical pathway complement–mediated hemolytic assay.”
In vitro classical pathway convertase activity assay
The activity of the classical pathway C4b2a in directly activating C5 was assessed following a previously published protocol with minor revisions.10,11 Briefly, EshA cells (2 × 107) were incubated with 30% C3 Dpl human sera in GVB++ buffer at 37°C for 5 minutes together with 5 μM of staphylococcal superantigen-like protein 7 (SSL7),12,13 a potent C5 inhibitor, to allow the formation of C4b2a without hemolysis. After incubation, EshA cells with the assembled C4b2a on the surface were washed and incubated with 100% WT or C3 KO rat sera in the presence of 10 mM EDTA for another 60 minutes. Activation of C5 directly by C4b2a was assessed by measuring MAC-mediated hemolysis.
In vivo clearance of RBC assay
A total of 3 × 109 EshA or Erabb cells were labeled by Dil cell-labeling solution (Thermo Fisher Scientific, Waltham, MA) and resuspended in 0.5 mL of phosphate-buffered saline. WT and C3 KO rats were injected IV with the labeled RBCs. Blood was collected from the tail vein at different time points (from 1 to 30 minutes), and the survival of infused RBCs was analyzed using a flow cytometer. Some of the infused rats were euthanized by using carbon dioxide 5 minutes later, and blood was collected by cardiac puncture to measure levels of released hemoglobin by reading OD414.
Results
Absence of C3 abolishes alternative pathway–mediated hemolysis in vitro
We initially compared hemolysis mediated through the alternative pathway of complement activation in vitro using Erabb cells incubated with 5% to 40% of either C3-Dpl human sera or C3 KO rat sera We observed that both depletion of C3 and deficiency in C3 nearly abolished hemolysis mediated by the alternative pathway of complement activation in both the human and rat complement systems in all tested concentrations of sera (Figure 1A-B). This conclusion remained true even when Erabb cells were incubated with 100% sera (Figure 1C). These data suggest that inhibiting C3 would be effective in significantly reducing alternative pathway–mediated tissue damage.
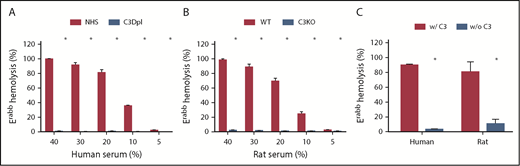
Comparison of the alternative pathway–mediated hemolysis between the C3-sufficient and C3-Dpl human or C3-deficient rat sera. For the measurements of alternative pathway–mediated hemolysis, Erabb cells were incubated with different concentrations of NHS ranging from 5% to 40%, C3-Dpl human sera (A) or WT C3-deficient (C3KO) rat sera (B) in GVB-Mg++/EGTA buffer, and hemolysis was quantitated by measuring levels of released hemoglobin in the supernatants. (C) In some experiments, Erabb cells were incubated with 100% human or rat sera, and hemolysis was assessed by following the same protocol. These data show almost complete abolition of hemolysis in the C3-Dpl human sera and C3-deficient rat sera. *P < .05.
Comparison of the alternative pathway–mediated hemolysis between the C3-sufficient and C3-Dpl human or C3-deficient rat sera. For the measurements of alternative pathway–mediated hemolysis, Erabb cells were incubated with different concentrations of NHS ranging from 5% to 40%, C3-Dpl human sera (A) or WT C3-deficient (C3KO) rat sera (B) in GVB-Mg++/EGTA buffer, and hemolysis was quantitated by measuring levels of released hemoglobin in the supernatants. (C) In some experiments, Erabb cells were incubated with 100% human or rat sera, and hemolysis was assessed by following the same protocol. These data show almost complete abolition of hemolysis in the C3-Dpl human sera and C3-deficient rat sera. *P < .05.
Absence of C3 does not prevent classical pathway–mediated hemolysis in vitro
We then compared hemolysis mediated through the classical pathway of complement activation in vitro using EshA cells incubated with NHS, C3-depleted human sera, or WT C3 KO rat sera. Surprisingly, we observed reduced RBC damage in experiments analyzing the classical pathway–mediated hemolysis only when lower concentrations of human (up to 30%) or rat (up to 2%) sera were used (Figure 2A-B). At higher serum concentrations (>30% for humans; >5% for rats), both NHS and C3-Dpl human sera as well as WT and C3 KO rat sera induced comparable and almost complete classical pathway–mediated hemolysis (Figure 2C-D).
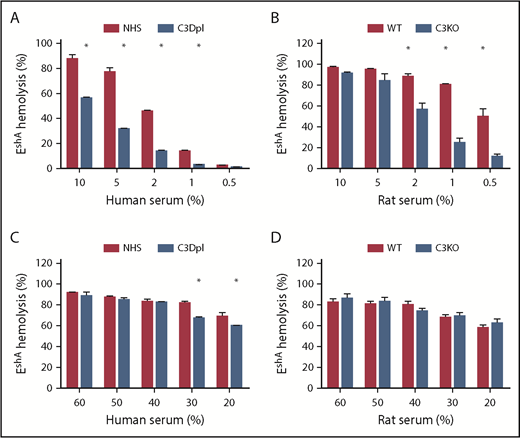
Comparison ofthe classical pathway–mediated hemolysis between the C3-sufficient and C3-Dpl human or C3-deficient rat sera. For the measurements of classical pathway–mediated hemolysis, antibody-sensitized EshA cells were incubated with different concentrations of NHS, C3-Dpl human sera (A,C), or WT C3-deficient (C3KO) rat sera (B,D) in GVB++ buffer, and hemolysis was quantitated by measuring levels of released hemoglobin in the supernatants. These data show significantly reduced classical pathway–mediated hemolysis in the absence of C3 when lower concentrations of sera were used (A-B), but comparable and almost complete hemolysis when higher concentrations of sera were incubated (C-D). *P < .05
Comparison ofthe classical pathway–mediated hemolysis between the C3-sufficient and C3-Dpl human or C3-deficient rat sera. For the measurements of classical pathway–mediated hemolysis, antibody-sensitized EshA cells were incubated with different concentrations of NHS, C3-Dpl human sera (A,C), or WT C3-deficient (C3KO) rat sera (B,D) in GVB++ buffer, and hemolysis was quantitated by measuring levels of released hemoglobin in the supernatants. These data show significantly reduced classical pathway–mediated hemolysis in the absence of C3 when lower concentrations of sera were used (A-B), but comparable and almost complete hemolysis when higher concentrations of sera were incubated (C-D). *P < .05
In addition to the above-described, well-established assays using xenogenic EshA cells to assess the classical pathway–mediated hemolysis (tissue damage), we also used a syngeneic experimental system in which we first prepared antibody-sensitized rat RBCs and then incubated them with different concentrations of WT or C3 KO rat sera, followed by hemolysis comparison. Similar to the results obtained using EshA cells, these studies using syngeneic rat RBCs and rat sera showed that even though there was significantly reduced hemolysis in the absence of C3 when lower concentrations of sera were used (<50%), at higher concentrations of complement (sera; >50%), the C3 deficiency in the sera did not protect the antibody-sensitized RBCs from complement-mediated hemolysis at all (Figure 3A).
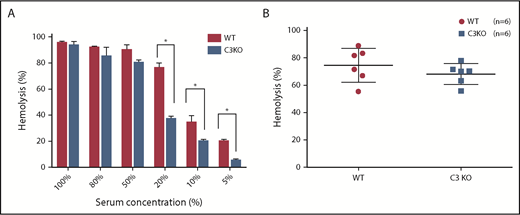
Analyses ofthe classical pathway–mediated hemolysis in a syngeneic experimental system using preassembled C4b2a. (A) For measuring classical pathway–mediated hemolysis in a syngeneic experimental system, antibody-sensitized rat RBCs were prepared using an anti-rat antiserum, then incubated with different concentrations of WT or C3-deficient (C3KO) rat sera in GVB++ buffer, and hemolysis was quantitated by measuring levels of released hemoglobin in the supernatants. These data show significantly reduced classical pathway–mediated hemolysis in the absence of C3 when lower concentrations of sera were used but comparable and almost complete hemolysis when higher concentrations of sera (50%-100%) were incubated. (B) In other experiments, the classical pathway C3 convertases were assembled on EshA cells after incubation with C3-Dpl sera in the presence of the potent C5 inhibitor SSL7 to suppress hemolysis. Then the EshA cells with preassembled C4b2a were incubated with sera from different WT and C3 KO rats (n = 6 in each group). Hemolysis was measured to assess the activities of C4b2a in directly activating C5. These results show that the preassembled C4b2a caused almost completely hemolysis in the presence and absence of C3, demonstrating that C4b2a can directly activate C5 in the absence of C3. *P < .05.
Analyses ofthe classical pathway–mediated hemolysis in a syngeneic experimental system using preassembled C4b2a. (A) For measuring classical pathway–mediated hemolysis in a syngeneic experimental system, antibody-sensitized rat RBCs were prepared using an anti-rat antiserum, then incubated with different concentrations of WT or C3-deficient (C3KO) rat sera in GVB++ buffer, and hemolysis was quantitated by measuring levels of released hemoglobin in the supernatants. These data show significantly reduced classical pathway–mediated hemolysis in the absence of C3 when lower concentrations of sera were used but comparable and almost complete hemolysis when higher concentrations of sera (50%-100%) were incubated. (B) In other experiments, the classical pathway C3 convertases were assembled on EshA cells after incubation with C3-Dpl sera in the presence of the potent C5 inhibitor SSL7 to suppress hemolysis. Then the EshA cells with preassembled C4b2a were incubated with sera from different WT and C3 KO rats (n = 6 in each group). Hemolysis was measured to assess the activities of C4b2a in directly activating C5. These results show that the preassembled C4b2a caused almost completely hemolysis in the presence and absence of C3, demonstrating that C4b2a can directly activate C5 in the absence of C3. *P < .05.
Preassembled classical pathway C3 convertase C4b2a directly activates C5 to cause hemolysis
Our results described in “Absence of C3 does not prevent classical pathway–mediated hemolysis in vitro” using C3-Dpl human sera and rat C3 KO sera suggest that the classical pathway C3 convertase C4b2a directly activates C5, leading to MAC formation and hemolysis. To test this hypothesis, we assembled C4b2a on EshA cells by incubating EshA cells with C3-Dpl human sera by following a previously published protocol.10,11 We had to add the potent C5 inhibitor SSL712,13 to the system to suppress hemolysis while allowing the formation of C4b2a. After washing, we incubated the EshA cells with preassembled C4b2a with either WT or C3 KO rat sera in the presence of EDTA and then measured MAC-mediated hemolysis. These experiments showed that the preassembled C4b2a directly caused hemolysis when WT or C3 KO rat sera was used (Figure 3B), demonstrating that the classical pathway C3 convertase (C4b2a) can directly activate C5 in the absence of C3 to cause hemolysis.
Absence of C3 significantly reduced alternative pathway–mediated hemolysis in vivo
To validate the in vitro results in vivo, we first infused identical numbers of fluorescence-labeled Erabb cells into WT or C3 KO rats via tail vein injection, subsequently bled the rats at different time points, and evaluated the survival of the infused RBCs by flow cytometry. We also euthanized some of the rats 5 minutes after infusion to collect blood by cardiac puncture for quantitating levels of released hemoglobin in the sera. Consistent with the in vitro hemolysis results, the C3 KO rats infused with rabbit RBCs had significantly longer RBC survival (Figure 4A) and minimal and markedly lower serum hemoglobin levels than did the WT rats (Figure 4B).

Comparison of complement-mediated hemolysis in WT and C3 KO rats. Identical numbers of labeled Erabb or EshA cells were IV infused into WT or C3 KO rats, and the survival of these labeled RBCs at different time points was monitored by flow cytometry. These data show rapid and comparable kinetics of the clearance of the infused EshA cells (C) but significantly prolonged survival of the infused Erabb cells in the C3 KO rats (n = 6 in each group) (A). The in vivo experiments were repeated, and this time the rats were euthanized 5 minutes after infusion to collect blood by cardiac puncture for the comparison of levels of serum hemoglobin. These data show that compared with the naïve controls (without any RBC infusion), both the WT and C3 KO rats that received EshA cell infusion had significantly increased levels of serum hemoglobin with even higher levels in the C3 KO rats than in the WT rats. (D) However, levels of serum hemoglobin in the Erabb cell infused C3 KO but not WT rats were significantly reduced and comparable to those in the untreated rats (control) (B) (n = 4 in each group). *P < .05.
Comparison of complement-mediated hemolysis in WT and C3 KO rats. Identical numbers of labeled Erabb or EshA cells were IV infused into WT or C3 KO rats, and the survival of these labeled RBCs at different time points was monitored by flow cytometry. These data show rapid and comparable kinetics of the clearance of the infused EshA cells (C) but significantly prolonged survival of the infused Erabb cells in the C3 KO rats (n = 6 in each group) (A). The in vivo experiments were repeated, and this time the rats were euthanized 5 minutes after infusion to collect blood by cardiac puncture for the comparison of levels of serum hemoglobin. These data show that compared with the naïve controls (without any RBC infusion), both the WT and C3 KO rats that received EshA cell infusion had significantly increased levels of serum hemoglobin with even higher levels in the C3 KO rats than in the WT rats. (D) However, levels of serum hemoglobin in the Erabb cell infused C3 KO but not WT rats were significantly reduced and comparable to those in the untreated rats (control) (B) (n = 4 in each group). *P < .05.
Absence of C3 did not significantly reduce classical pathway–mediated hemolysis in vivo
To test the significance of C3 deficiency in preventing classical pathway complement–mediated hemolysis in vivo, we infused identical numbers of fluorescence-labeled EshA cells into WT or C3 KO rats via tail vein injection. We again subsequently bled the rats at different time points to evaluate the survival of the infused RBCs by flow cytometry and euthanized some of the rats 5 minutes after infusion to collect blood by cardiac puncture to quantitate levels of released hemoglobin in the sera. Notably, both the WT and C3 KO rats infused with antibody-sensitized sheep RBCs showed rapid clearance of the RBCs with comparable kinetics (Figure 4C) and significant levels of serum hemoglobin in the blood. Interestingly, levels of released hemoglobin are even higher in the C3 KO rats than in the WT rats (Figure 4D).
Discussion
In this study, using established hemolysis assays, we found that neither C3-Dpl human sera nor C3 KO rat sera could lyse Erabb cells, but both C3-Dpl human and C3 KO rat sera efficiently lysed EshA cells or antibody-sensitized rat RBCs. Further experiments using preassembled classical pathway convertase C4b2a also confirmed that this enzyme directly activated C5 to cause hemolysis. Results from in vivo studies using WT and C3 KO rats were consistent with those from the in vitro experiments, showing that although Erabb cells were significantly protected in C3 KO rats, EshA cells were not. All these results suggest that C3 is not essential for classical pathway–mediated hemolysis; therefore, inhibiting C3 might not be effective in preventing classical pathway–mediated tissue damage.
In our studies, the RBCs were lysed by MACs that assembled on the cell surfaces. MAC assembly requires the C3 convertases and C5 convertases (C3bBb and C3bBbC3b) in the alternative pathway to activate C3 and C5, respectively. The status of C3 as an essential component of both C3 and C5 convertases in the alternative pathway might explain the near-complete abolition of hemolysis after incubating rabbit RBCs with C3-Dpl human sera or C3 KO rat sera. By contrast, the C3 convertase in the classical pathway of complement activation (C4b2a) can form in the absence of C3. Although the formation of the C5 convertase in the classical pathway (C4b2aC3b) still requires C3, previous studies by others demonstrated that C5 can still be directly activated by the C3 convertase C4b2a with reduced efficacy,14,15 which triggers the assembly of MAC and, consequently, the lysis of RBCs. Indeed, our experiments using preassembled C4b2a and WT or C3 KO rat sera also demonstrated that the classical pathway C3 convertase directly activated C5 without the requirement of C3, leading to hemolysis. Because the lectin pathway shares the same C3 and C5 convertases as the classical pathway, it is also expected that these results apply to the lectin pathway of complement activation.
Our in vitro studies limited RBC lysis to intravascular hemolysis because the experimental system contained only complement but lacked phagocytes. Because C3b/iC3b opsonization of RBCs16,17 and Fc receptors18 on phagocytes both facilitate phagocytosis, extravascular and intravascular hemolysis can occur simultaneously in vivo. Consistent with the in vitro results derived using human and rat sera in the presence or absence of C3, the in vivo results demonstrated no difference in the clearance of antibody-sensitized RBCs in the WT and C3 KO rats, as measured by the survival of the infused RBCs. There was even a significantly increased level of released hemoglobin in the C3 KO rats compared with the WT rats, likely as a result of reduced phagocytosis in the absence of C3, leading to an increased proportion of the hemolysis occurring in the intravascular compartment and higher levels of serum hemoglobin as a result of hemolysis. These results suggest that C3 should be an effective target in the inhibition of alternative pathway–mediated hemolysis. This is consistent with positive results from a clinical trial using a C3 inhibitor for treating PNH,19,20 in which the alternative pathway of complement activation plays a primary role in the pathogenesis.21,22 However, C3 inhibition might not be a valid approach toward reducing intravascular hemolysis when the classical pathway (and in principle, the lectin pathway) of complement activation plays a major role in the process, even though extravascular hemolysis could be significantly reduced. These data might also explain the recent positive preliminary results from a clinical trial using a C3 inhibitor for treating CAD23 in which complement activation is initiated by the antibody-antigen complexes on the RBCs, but extravascular hemolysis is the predominant route of RBC damage in patients.24
The importance of the alternative pathway in complement-mediated pathological conditions has been demonstrated in many human and animal studies.25 It is a pathway to initiate complement activation and also to amplify complement activation initiated by other pathways (eg, the classical and lectin pathways). Even though our results using C3-Dpl human sera and C3 KO rats suggest that targeting C3 might not be effective for preventing classical pathway–mediated hemolysis, C3 inhibitors might still be beneficial in conditions in which complement activation is initiated by the classical (lectin) pathway, but the amplification of the activated complement activation plays an integral role in the pathogenesis.
Collectively, we have studied complement-mediated hemolysis both in vitro and in vivo using NHS and C3-Dpl human sera, as well as WT and C3 KO rats. Our results suggest that C3 inhibition should effectively reduce alternative pathway–mediated hemolysis, and by implication, tissue damage. However, this approach might not similarly affect classical pathway–mediated tissue damage. These results provide new insights into the potential for the use of C3-targeted therapeutics for the treatment of complement-mediated diseases.
Acknowledgments
This project is supported in part by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant R01 DK103581 (F.L.). Y.D. is supported in part by West China Hospital, Chengdu, China.
Authorship
Contribution: L.Z., Y.D., and P.H. performed the experiments; T.L.S. and D.A.F. helped develop the C3 KO rats, discussed the results, and edited the manuscript; J.X. discussed the results and edited the manuscript; and F.L. designed the experiments and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Feng Lin, Department of Inflammation and Immunity, Lerner Research Institute, Cleveland Clinic, 9500 Euclid Ave, Cleveland, OH 44195; e-mail: linf2@ccf.org.

