


Abstract
Intensive scrutiny of human genomes has unveiled considerable genetic variation in coding and noncoding regions. In cancers, including those of the hematopoietic system, genomic instability amplifies the complexity and functional consequences of variation. Although elucidating how variation impacts the protein-coding sequence is highly tractable, deciphering the functional consequences of variation in noncoding regions (genome reading), including potential transcriptional-regulatory sequences, remains challenging. A crux of this problem is the sheer abundance of gene-regulatory sequence motifs (cis elements) mediating protein-DNA interactions that are intermixed in the genome with thousands of look-alike sequences lacking the capacity to mediate functional interactions with proteins in vivo. Furthermore, transcriptional enhancers harbor clustered cis elements, and how altering a single cis element within a cluster impacts enhancer function is unpredictable. Strategies to discover functional enhancers have been innovated, and human genetics can provide vital clues to achieve this goal. Germline or acquired mutations in functionally critical (essential) enhancers, for example at the GATA2 locus encoding a master regulator of hematopoiesis, have been linked to human pathologies. Given the human interindividual genetic variation and complex genetic landscapes of hematologic malignancies, enhancer corruption, creation, and expropriation by new genes may not be exceedingly rare mechanisms underlying disease predisposition and etiology. Paradigms arising from dissecting essential enhancer mechanisms can guide genome-reading strategies to advance fundamental knowledge and precision medicine applications. In this review, we provide our perspective of general principles governing the function of blood disease–linked enhancers and GATA2-centric mechanisms.
Introduction
Representing a human genetics revolution, next-generation DNA sequencing is routinely used in clinical settings to obtain patient-specific insights into disease etiology, progression, and drug sensitivity. Typically, DNA sequences of exons from a limited candidate gene cohort (panel) are analyzed. Alternatively, whole-exome sequencing generates sequences from a much larger gene cohort. Standardized algorithms are deployed to distinguish between innocuous genetic variation and variation that informs clinical medicine. Simultaneously assessing the structural integrity of many protein-coding genes has been transformative. From the perspective of transitioning outside of known territory, however, a major limitation is that these analyses are blind to sequences beyond exons at enhancers, promoters, and chromatin insulators. Genetic variation in noncoding sequences is commonly deemed “variants of undetermined significance.” Because cis-element genetic variation can yield phenotypic consequences as profound as null mutations within a gene, panels and exome sequencing yield incomplete sketches with intrinsic limitations for advancing genome science and patient care.
The shortcomings of gene panels and whole-exome sequencing can be surmounted by whole-genome sequencing, albeit the cost can be prohibitive in clinical contexts. Irrespective of economics, whole-genome sequencing fails to detect or discards sequences from genomic regions with physical properties that create obstacles to sequencing analytical pipelines, for example, repetitive sequences that do not map uniquely to discrete targets. Repetitive sequences, such as retrotransposons, can confer regulatory functions.1 In pathologies characterized by a low mutant allele burden, mutation detection necessitates a high sequencing depth. Irrespective of obstacles to documenting variation, it remains challenging to definitively ascertain the significance of noncoding region variation. From an acute clinical perspective, the less than optimal genome-reading logistics may yield data that are not deemed beneficial and/or generate more questions than answers. Whole-genome–sequencing data can influence perceptions regarding a patient’s health and/or propensity to develop disease, even though the data may not yield high-fidelity predictions nor inform interventions. Although we have only begun to scratch the surface of deciphering noncoding sequence variation, as genome-reading acumen improves, clinically annotated patient-sequence banks will constitute an invaluable resource to advance fundamental and clinical/translational research.
An attractive approach for deciphering genome function involves amalgamating genomic data documenting histone posttranslational modifications, cytosine guanine dinucleotide DNA methylation and hydroxymethylation, chromatin accessibility, transcription factor and coregulator occupancy with evolutionary conservation, and DNA sequence to generate topographic maps genome-wide.2 This limited-dimensional analysis can be enhanced via strategies that incorporate 3-dimensional chromosome conformation, for example, HiC3 and Capture C,4,5 to reveal the spatial relationship between a putative regulatory sequence and neighboring genes that may not be evident in 2-dimensional space.6 Genetic variation may not necessarily impact the nearest-neighbor genes in 2 dimensions. To pinpoint bona fide regulatory elements, combinations of these parameters can yield instructive predictions.7-11 Inferences regarding potential functionality require direct testing, which is enabled by gene-editing technologies, with zinc-finger nucleases12,13 or transcription activator-like effector nucleases (TALENs)14 and now predominantly clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR–associated protein 9 (Cas9)15-17 or Cas9-like permutations18 to excise sequences from a genome. Alternatively, one can use designer fusion proteins containing a DNA-binding domain recognizing the sequence of interest fused to a module that activates or represses genes at the docking site.19-21 Although rigorous functional analyses at endogenous loci are increasingly feasible, they remain challenging in low-abundant cell populations and contexts that cannot be recapitulated faithfully with cultured cells. Given the swift pace of this technology development, current limitations will likely be surmounted in the near-term, and overcoming difficulties will further transform genome science, clinical genetics, and precision medicine.
Establishing and maintaining cell-type–specific transcriptomes
As genomes generate dynamically regulated transcriptomes, the transcriptome, which can be straightforward to measure, serves as an invaluable proxy of genome functional status. Cell-type–specific transcription factors act in concert with large ensembles of broadly expressed transcription factors and coregulators at enhancers to establish and maintain transcriptomes. Enhancers were discovered as DNA sequences that confer position-independent and orientation-independent expression of genes on plasmids transfected into cells.22 As technologies evolved from plasmids to transgenes integrated at ectopic chromosomal sites and ultimately endogenous loci, it became clear that enhancers can reside within introns or quite far (eg, 100 kb) or close to a gene. Enhancer function in plasmids often does not correlate, quantitatively or qualitatively, with endogenous locus function. Enhancers can activate reporter genes in plasmids hundreds of fold, for example, the β-globin hypersensitive site II enhancer,23 while contributing incrementally (eg, ∼20%) to endogenous locus activity in vivo.24,25 Enhancers consist of clustered cis elements mediating transcription factor binding, and their sizes range from that approximating nucleosomal DNA (200 bp)26,27 to several or many kilobases (superenhancer28 ). Regardless of whether an enhancer is remote or proximal to a gene and small or large, enhancer-bound transcription factors recruit chromatin-modifying and -remodeling coregulators29 and RNA polymerase II.30,31 Through enzymatic functions, in which coregulators posttranslationally modify histones, and interactions with transcription factors, enhancers stimulate higher-order chromatin transitions (looping)32,33 ; whether sustained or transient enhancer-promoter interactions are essential is still debated.34,35 Regardless of sustained vs transient looping, enhancers relocalize loci within the 3-dimensional topography of the nucleus and its functionally distinct subdomains.36-38
Knowledge on sequence and chromatin attributes of enhancers has led to strategies to predict enhancers genome-wide. One or more of the following have enhancer-predictive utility, at least for activity in transfection assays and upon transgene integration at ectopic loci: elevated histone H4 monomethylated at lysine 4, histone H3 acetylated at K27, p300 occupancy, chromatin accessibility, higher-order chromatin conformation, enhancer-derived RNAs, and evolutionary conservation.39-42 Whether these parameters can be used broadly to predict activity at endogenous loci is unclear. Much more importantly is discriminating between essential enhancers exerting critical functions vs modulatory or redundant enhancers not vital for cellular and organismal functions. However, even a modest degree of enhancer activity may control a crucial biological process.
Examples have emerged in which a sequence fulfills the criteria to qualify as an enhancer, having activity in plasmids and/or in transgenes, yet its excision from the genome yields little to no impact on expression of the associated gene.24,25,43 In this case, the enhancer might target a gene residing in proximity in 3- but not 2-dimensional space, reinforcing the need to consider chromosomal conformation. Significant caveats with reconstructing 3-dimensionality include the reliance on formaldehyde to crosslink macromolecules in cells, which yields false-positives and incompletely traps conformations. Conformational maps may only inform the specific cellular contexts in which they are generated, given the genome remodeling intrinsic to cellular processes such as differentiation. The 3-dimensional configuration of loci in erythroleukemia cells has limited utility to inform genome function in hematopoietic progenitor cells. Ascertaining functional implications of an enhancer deletion requires global transcriptional profiling to establish whether the deletion impacts genes proximal to the enhancer and genes predicted to reside in the neighborhood, based on chromatin conformation. Of course, if an enhancer controls expression of a transcription factor, for example, GATA2, many genes will be dysregulated indirectly. Enhancer-dependent cellular phenotypes can also be highly informative. If an enhancer deletion does not influence neighboring genes or genes more broadly and does not elicit cellular phenotypes, either the enhancer is not essential to regulate the neighboring genes, it regulates genes not critical for cellular physiology, or the physiological processes regulated are not recapitulated in the system or are unknown. A negative result may also reflect redundant activity masked by other cis elements, nonredundant activity in particular cell types, or contexts distinct from those analyzed or misassignment of the sequence, which is not a bona fide enhancer.
Although the number of enhancers analyzed at endogenous loci in vivo is increasing, reports of those demonstrated unequivocally to exert essential activities (eg, required for development or others vital processes) are limited. It is unclear whether most enhancers function in vivo as all-or-none switches to convert repressed into active genes, or whether a spectrum of activities exists ranging from switch-like behavior to modulatory adjustments in gene expression, the latter being more difficult to analyze and interpret.
Discovering enhancers essential for hematopoiesis: GATA2 paradigm
Because enhancer activities can be highly context-dependent, it is instructive to consider how they establish/maintain unique protein expression patterns, for example, in stem and progenitor cells. It is not our intent to describe all enhancers studied in the hematopoietic system, but rather to focus on principles illustrated by essential enhancers that control transcription and important biological processes.
GATA2 is required for hematopoietic stem cell (HSC) generation and function,44-47 myeloerythroid progenitor generation and function,48,49 the function of committed erythroid precursors,49 and even endothelial cell function.50-57 As disrupted expression or mutational alteration of GATA2 are pathogenic, ensuring the fidelity of GATA2 expression and function in these distinct cellular contexts is crucial. GATA2 expression in hemogenic endothelial cells in the aorta gonad mesonephros region (AGM) of the embryo induces HSC emergence.54,58 GATA2 expression in HSCs confers long-term repopulating activity, and its expression in myeloerythroid progenitors and erythroid precursors confers differentiation potential.45,48,49,59 GATA2 stimulates cellular proliferation and promotes survival,44,47,60 although the underlying mechanisms, and whether unifying mechanisms operate in the distinct contexts, are unclear. As GATA2 functions in widely variable regulatory milieus, mechanisms governing its expression are likely to be context-dependent. Alternatively, a comparable cohort of regulatory factors in distinct contexts might generate a common mechanism.
Gata2 nucleoprotein structure was initially elucidated in a mouse erythroid precursor cell line lacking GATA1 (G1E)61,62 that expresses endogenous GATA2. This work led to the discovery of conserved Gata2 enhancers with essential activities to control embryogenesis, as well as developmental and regenerative hematopoiesis.48,51,63-67 The +9.5 and −77 (9.5 and 77 kb downstream and upstream of the transcription start site) enhancers are essential for embryogenesis and hematopoiesis in vivo (Figure 1), and their disruption leads to human pathologies including leukemia.48,51,68 As GATA1 is required for erythroid and megakaryocytic differentiation,69-72 GATA2-expressing G1E cells propagate in an immature, erythroid precursor state. Activation of a conditional GATA1 allele (ER-GATA1) represses Gata2 transcription and induces erythroid maturation.63,73 GATA1 replaces GATA2 at 5 Gata2 sites.74-76 These GATA switches correlate with repression, suggesting that GATA2 positively autoregulates Gata2, and GATA1 represses Gata2, in part, by disrupting positive autoregulation. Reciprocal GATA2 and GATA1 expression patterns occur in diverse mouse and human erythroid systems.77-79
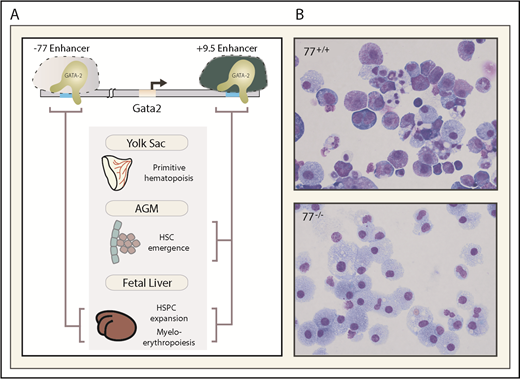
Essential enhancers governing Gata2 expression and hematopoiesis. (A) The −77 and +9.5 enhancers reside 77 and 9.5 kb upstream and downstream, respectively, of the Gata2 transcriptional start site. These evolutionarily conserved enhancers control mouse and human Gata2 transcription in specific biological contexts and vital steps in hematopoiesis (depicted with brackets). (B) The photomicrographs depict Giemsa-stained myeloerythroid progeny resulting from ex vivo differentiation of −77+/+ or −77−/− fetal liver progenitor cells. AGM, aorta gonad mesonephros region of the embryo proper.
Essential enhancers governing Gata2 expression and hematopoiesis. (A) The −77 and +9.5 enhancers reside 77 and 9.5 kb upstream and downstream, respectively, of the Gata2 transcriptional start site. These evolutionarily conserved enhancers control mouse and human Gata2 transcription in specific biological contexts and vital steps in hematopoiesis (depicted with brackets). (B) The photomicrographs depict Giemsa-stained myeloerythroid progeny resulting from ex vivo differentiation of −77+/+ or −77−/− fetal liver progenitor cells. AGM, aorta gonad mesonephros region of the embryo proper.
In vitro studies suggested that 1 or more of the Gata2 GATA switch sites might establish Gata2 transcription in vivo and/or GATA1-instigated Gata2 repression during erythroid maturation. These possibilities were tested using mice lacking the individual sites. Individual deletions of sites −1.8, −2.8, and −3.9 kb relative to the Gata2 promoter had little to no consequences for Gata2 expression, hematopoiesis, and stress erythropoiesis, yet these sites exhibited enhancer attributes.43,66,67 As the −1.8-kb deletion resulted in Gata2 upregulation in erythroblasts where Gata2 is normally repressed, this site was required for maintenance, but not initiation, of Gata2 repression.66 The −2.8-kb deletion modestly reduced Gata2 expression in progenitors, suggesting its enhancer function is modulatory, rather than a critical switch.67 The −3.9 deletion had little to no impact on Gata2 expression and hematopoiesis.43
Unlike the −1.8, −2.8, and −3.9 deletions that removed conserved GATA motifs and neighboring sequences representing potential cis elements, a 46-bp deletion of the intronic +9.5 site was lethal at approximately embryonic day 13.5 (E13.5).51 This contrasts with approximately E10.5 lethality of the Gata2 coding region knockout.44 The +9.5 deletion abrogated HSC emergence in the AGM and strongly reduced fetal liver hematopoietic stem and progenitor cells (HSPCs).51,58 As the mutant embryos retained abundant primitive erythroid cells, and E9.5 yolk sac generated primitive erythroid colonies with no obvious defects ex vivo, the +9.5 deletion selectively impairs definitive hematopoiesis.51 Although +9.5 intronic localization differs from the −1.8, −2.8, and −3.9 sites, the sites shared GATA factor occupancy, variable degrees of enhancer activity in transfection assays, and enhancer-predictive chromatin attributes. The +9.5 constitutes the sole report of an enhancer essential for triggering stem cell generation.
An evolutionarily conserved GATA factor-occupied sequence (−77) resides downstream of Rpn1, the nearest neighbor to the Gata2 5′ end. As GATA1 represses Gata2 transcription and does not regulate Rpn1 expression,63 we hypothesized that −77 is a distal enhancer that controls Gata2, collectively with +9.5 or independently in specific contexts.65,74 Like the +9.5, a 257-bp deletion of the −77 is embryonic lethal, but −77−/− embryos live longer (lethality after approximately E15.5) than +9.5−/− embryos.48 The −77 mutant fetal liver myeloerythroid progenitors retain the capacity to undergo monocytic differentiation, while erythroid and granulocytic differentiation is nearly abrogated.48,49 Despite the essential +9.5 requirement for HSC emergence in the AGM, and essential +9.5 and −77 functions, HSC emergence and HSC activity during embryogenesis are unaffected in −77−/− embryos.48 Consistent with this activity, −77 confers Gata2 expression in progenitors, but not in multipotential precursors. Thus, 2 conserved Gata2 enhancers control distinct processes: +9.5-regulated HSC emergence and −77-regulated progenitor fate.
It is unknown whether sequences extending beyond the core contribute to core activity. As superenhancers are large enhancers operating under what appear to be similar principles to enhancers (transcription factor occupancy, coregulator recruitment, chromatin looping, etc), this designation does not uniquely inform mechanisms.
Because the +9.5, but not the −77, triggers HSC emergence,58,80 it was unclear whether the 2 enhancers ever function collectively. This problem was addressed by analyzing genetic interactions between heterozygous enhancer alleles at distinct anatomical sites and developmental stages. A compound heterozygous mutation eliminating 1 copy of each enhancer on distinct alleles (−77+/−;+9.5+/−) is lethal at approximately E14, with no impact on yolk-sac hematopoiesis and HSC emergence and function.49 Resembling −77−/− fetal liver myeloid progenitors, 77+/−;+9.5+/− progenitors generated predominantly macrophages ex vivo.49 The 77+/−;+9.5+/− fetal liver lacks megakaryocyte erythrocyte progenitors.49 Thus, both enhancers must reside on 1 allele to confer Gata2 expression to support progenitor function and megakaryocyte erythrocyte progenitor generation. The −77 and +9.5 do not interact genetically to control HSC emergence,49 illustrating an additional distinction between enhancer requirements for HSC generation vs progenitor generation/function. Interrogating genetic interactions between multiple enhancers at a locus has broad applicability to elucidate mechanisms operating in distinct contexts in vivo.
In aggregate, these analyses and others demonstrated that essential activities and their quantitative importance cannot be inferred from enhancer attributes conventionally used to predict “enhancers.” Multiple essential enhancers at the same locus can contribute qualitatively unique regulatory modes, and their integrated actions confer expression of GATA2, ensuring its capacity to govern HSPC transitions.
Leveraging essential enhancer attributes to discover enhancer cohorts genome-wide
Because general enhancer attributes do not yield high-fidelity predictions of essential enhancer activities at endogenous loci, can unique attributes of essential enhancers, for example, +9.5, be used to identify comparable enhancers? The +9.5 core conforms to an E-box (CATCTG) 8-bp spacer GATA motif (AGATAA) composite element55,56,81 (Figure 2). This configuration can confer GATA1- or GATA2-dependent enhancer activity in transfection assays,82-84 and multiple transcription factors (eg, Tal1 and Fli1) and coregulators (eg, Lmo2 and Ldb1) can co-occupy these motifs with the GATA factor.82,85,86 As the human genomes contain ∼8900 CATCTG 6- to 14-bp spacer AGATAA elements,87,88 it is instructive to consider what parameters render this sequence, in the context of +9.5, an essential enhancer. Is the composite element sufficient for factor binding and activity when situated in accessible chromatin? Do neighboring cis elements endow, amplify, or attenuate composite element activity? Does the location relative to gene features (eg, promoter, intron, exon, or distal) dictate activity? Does conservation discriminate functional from nonfunctional elements?

Mouse and human +9.5 enhancer alleles. The wild-type mouse +9.5 configuration is depicted, with 3 mutant mouse alleles below. Ets-only and GATA-only mutants fail to support developmental hematopoiesis and are both embryonic lethal. The Ets motif point mutant that models the human single-nucleotide mutation exhibits largely normal developmental hematopoiesis yet is incompetent to support regeneration of the hematopoietic system postmyeloablation. Human alleles identified in patients with GATA2-deficiency syndrome are depicted. Black arrow extending from +9.5 to promoter, transcriptional activation; gray arrow, partial activation; absence of an arrow, severely attenuated enhancer activity. E, E-box-binding protein.
Mouse and human +9.5 enhancer alleles. The wild-type mouse +9.5 configuration is depicted, with 3 mutant mouse alleles below. Ets-only and GATA-only mutants fail to support developmental hematopoiesis and are both embryonic lethal. The Ets motif point mutant that models the human single-nucleotide mutation exhibits largely normal developmental hematopoiesis yet is incompetent to support regeneration of the hematopoietic system postmyeloablation. Human alleles identified in patients with GATA2-deficiency syndrome are depicted. Black arrow extending from +9.5 to promoter, transcriptional activation; gray arrow, partial activation; absence of an arrow, severely attenuated enhancer activity. E, E-box-binding protein.
Hewitt et al88 identified all “+9.5-like” composite elements in mouse and human genomes and devised a multifactorial prioritization scheme to parse these potential cis elements using parameters characteristic of +9.5, including intronic localization, conservation, GATA2 occupancy, and chromatin attributes. Chromatin immunoprecipitation (ChIP) sequencing data sets (76 histone modification and 38 chromatin occupancy) were used to rank 797 +9.5-like elements, based on their +9.5-like molecular signature. The advent of genetic-editing technologies has transformed our ability to discriminate between potentially important vs essential enhancers. High- and low-ranked +9.5-like sequences were analyzed using TALENs to excise several elements from their endogenous loci, which identified functional GATA2-activated enhancers; the data set almost certainly harbors many more that remain to be validated.88 Deletion of an intronic composite element at the poorly studied Samd14 gene strongly reduces Samd14 expression in GATA2-expressing G1E cells, mouse bone marrow, and spleen.88,89 Mice lacking this enhancer revealed a Samd14 function to control erythrocyte regeneration.89 Phenylhydrazine- or phlebotomy-induced hemolytic anemia activated the enhancer, increasing Samd14 expression, stem cell factor–dependent c-Kit signaling, and erythrocyte regeneration. This response confers survival in anemia, thus linking an enhancer mechanism to a vital regenerative process.90
Although the strategy described in the prior paragraph is broadly applicable to identify essential enhancers, the exact combination of parameters that enables universal predictions is unknown. The parameters may be context-dependent. For example, in an embryonic stem cell, in which chromatin differs greatly from a differentiated cell, the attributes with enhancer-predictive utility might not extrapolate to all systems. Similarly, examples exist in which factor occupancy of chromatin has a propensity to occur at distal sites in 1 context and promoters in another.91 Thus, genomic location might constitute a parameter with context-dependent predictive utility.
Recently innovated high-throughput technologies offer new tools, when combined with rigorous locus-specific functional analyses, to identify essential enhancers. HiChIP involves trapping higher-order chromatin interactions in a cell, followed by ChIP to define factor occupancy at chromatin segments engaged in long-range interactions.92 By mapping H3K27ac, this approach yields insights into the proximity of potential enhancers to potential target genes.93 Strategies have deployed guide RNAs to direct recruitment of a Kruppel-associated box repressor domain fusion to catalytically inactive Cas9 to chromatin (CRISPR interference).19,20,94-96 Gasperini et al used this strategy to analyze functional consequences at 5920 candidate enhancers, profiling >250 000 single-cell transcriptomes, which revealed 470 “enhancer-gene pairs.”96 However, gene repression or activation by an artificial fusion protein may occur through diverse mechanisms, including those independent of disrupting endogenous enhancer activity. Even if altered expression reflects disrupted enhancer activity, whether the enhancer is essential, modulatory, or redundant in physiological and pathological contexts requires genome deletion analysis, ideally in vivo to interrogate developmental and context-dependent activities. Combining these approaches with single-nucleotide polymorphisms and genetic variation in pathological contexts can uncover enhancers linked to human phenotypes and disease.
Enhancer mechanisms that suppress nonmalignant and malignant blood diseases
Steven Holland at the National Institute of Allergy and Infectious Diseases/National Institutes of Health (NIH), one of the discoverers of germline GATA2-coding mutations in patients with immunodeficiency, myelodysplastic syndrome, and acute myeloid leukemia (AML; GATA2-deficiency syndrome),97-102 identified a patient with telltale signs of GATA2-related disease, yet lacking GATA2-coding mutations. Sequencing revealed a heterozygous germline 28-bp deletion that disrupts the +9.5 E-box and upstream sequences51 (Figure 2). Four additional patients harbored a single-nucleotide C-T transition in an Ets motif 23 bp downstream of the 3′ end of WGATAA.68 As GATA2 messenger RNA is lower in patient mononuclear cells, and the Ets mutation impairs +9.5 activity in a transfection assay, a haploinsufficiency mechanism of pathogenesis was proposed.68
Several hundred adult and pediatric patients with GATA2 germline mutations have been described, with the single-nucleotide Ets motif mutation being the most common.103 Despite multiple conserved +9.5 sequences,51,80 patient mutations have been restricted to the Ets motif and the E-box/upstream sequence and have not been detected in another Ets motif upstream of the E-box, GATA motif, or other sequences. Panels and exome sequencing would not detect +9.5 mutations, and whole-genome sequencing is deployed only in limited clinical contexts. Given that +9.5 is a vital determinant of GATA2 regulation and its disruption creates a disease predisposition,51,68,80,103 medical centers (eg, NIH Clinical Center and University of Chicago) screen for +9.5 genetic variation.
As with GATA2-coding mutant patients, not all +9.5-mutant patients develop disease, and there is major variability in the disease onset age.101,104 These findings suggest a model in which GATA2 mutations create a disease predisposition insufficient for pathogenesis, which is strongly supported by modeling the Ets mutation in mice.80 Ets motif–mutant embryos develop normally, and the adult hematopoietic system in the steady state is normal, including a nearly indistinguishable multipotent hematopoietic precursor (Lin−Sca1+Kit+ [LSK] cell population) transcriptome vs wild-type cells. However, the mutants are hypersensitive to 5-fluorouracil, which ablates progenitors, forcing HSCs to regenerate the hematopoietic system. The mutation corrupts the regenerating LSK cell transcriptome, enhances 5-fluorouracil–induced bone marrow failure, impairs hematopoietic regeneration, and increases lethality. Structure/function analyses revealed that the +9.5 GATA motif is insufficient to support developmental hematopoiesis without the E-box and Ets motifs80 (Figure 2).
As the Ets motif mutation sensitizes the hematopoietic system to a secondary insult,80 it is instructive to consider the spectrum of insults that impact the mutant human hematopoietic system and whether a predisposition mutation increases the probability of secondary mutations. In principle, a range of genetic and environmental aberrations may trigger the pathogenic consequences of the “silent” GATA2 mutation. Although these triggers are not established, patients with germline GATA2 mutations can acquire somatic mutations105-109 constituting potential triggers, which was reviewed recently.103 The triggering mechanism(s) might reduce expression of the heterozygous wild-type allele below a critical threshold, alter function of GATA2-regulated genetic network components or impact processes operating in parallel with GATA2 mechanisms that govern HSPC generation/function.
GATA2 establishes and maintains complex genetic networks.48,49,51,58,75,80,87 Network functionality relies on intranetwork circuit integrity and circuit integration. Genetic and environmental aberrations can disrupt network integrity in a nearly infinite number of ways, and pathogenesis may not emerge from a predominant molecular aberration. This model extends the haploinsufficiency concept to loss-of-function (enhancer mutation) and gain-of-function (GATA2 overexpression or ectopic signaling that increases GATA2 activity) scenarios, both corrupting networks that regulate stem/progenitor cells. This new vision of GATA2-linked pathogenesis is supported by findings that GATA2-coding disease mutations are not strictly loss of function.110,111 In a genetic rescue assay in primary progenitor cells, mutants can retain activity or exert activity greater than GATA2 at select target genes.110 Although the mutants are defective in rescuing erythroid differentiation in progenitors with reduced GATA2 expression, they can retain or have exaggerated granulopoiesis-inducing activity.110
Analogous to GATA2, expression of Spi1 encoding the myeloid transcription factor PU.1, an Ets family member, must be tightly controlled to ensure normal hematopoiesis.112-117 Although GATA2 is not a determinant of PU.1 expression in progenitors,48,49 it may function with PU.1, positively or negatively, in certain contexts.113 PU.1 levels are regulated by an enhancer 14 kb upstream of the Spi1 promoter (upstream regulatory element [URE]).114,116,117 Unlike +9.5 and −77, the URE is not essential for survival during development and in adult mice.114 Homozygous deletion of the URE causes a large, but incomplete, decrease in PU.1 expression in bone marrow LSK cells and B220+ B cells. Spi1−/− mice develop hematopoietic defects, including B-cell lymphoproliferative syndrome, altered early thymocyte development, T-cell lymphoma, and AML. As PU.1 expression is higher in mutant vs wild-type DN1 T cells, this enhancer appears to have context-dependent repressor activity. Unlike +9.5 and −77, in which deletion phenotypes were ∼100% penetrant, URE deletion phenotypes vary considerably (eg, 6% to 64%). It was proposed that Wnt signaling targets the URE to induce Spi1 transcription, thereby generating lymphocyte progenitors, whereas differentiation-associated declines in Wnt signaling downregulate PU.1, facilitating T-cell specification.114 Heterozygous URE-mutant mice, in which PU.1 decreases by ∼35%, develop a preleukemia state, and combining this mutation in a Msh2−/− background yields AML.117 Msh2 encodes a DNA mismatch repair component. In humans, a URE single-nucleotide polymorphism impacts protein binding and reporter gene activity and is associated with an approximately twofold lower level of endogenous Spi1 expression. As disrupting DNA repair machinery triggers leukemogenesis, it will be instructive to assess whether this mechanism can be extrapolated to other predisposition mutations.
Because enhancers consist of clustered cis elements, and individual elements can be vital for activity, there is ample opportunity for mutational disruption or generation of transcription factor–binding sites within enhancers. Somatic mutations can create transcription factor–binding sites118 that activate or repress a neighboring gene. If such a change occurs at a chromatin site permissive for factor binding, this may dysregulate genes via multiple mechanisms. The ectopically bound transcription factor might induce assembly of a complex that activates a repressed gene, upregulates an expressed gene, alters expression dynamics, or attenuates transcription by displacing endogenous factors from adjacent or overlapping sites, diverting factors away from prescribed locations or creating inhospitable chromatin. Heterozygous somatic indels in T-cell acute lymphocytic leukemia (T-ALL) cell lines and patient samples generate Myb transcription factor–binding sites upstream of the T-ALL oncogene TAL1.118 Ectopic Myb occupancy correlates with occupancy by other transcription factors and acquisition of superenhancer attributes. A heterozygous single-nucleotide change ∼4 kb upstream of the oncogenic LMO1 locus generates a Myb transcription factor–binding site at a region lacking a known enhancer, which induces LMO1 overexpression in T-ALL.119 Somatic intronic indels at the T-ALL oncogene LMO2 elevate LMO2 expression.120 For somatic mutations in heterogeneous cell populations, it cannot be assumed that potentially deleterious alterations are critical, as sequence motifs in chromatin are often inaccessible to binding proteins. Discriminating between chromosomal aberrations with cancer-driving activity vs those merely reflecting genomic instability, and therefore surmounting a major impediment to cancer genome reading, necessitates detailed functional analyses.
Usurping enhancer function as a blood cancer–causing mechanism
As a paradigm-establishing discovery, a translocation links MYC-coding sequences to an immunoglobulin H (IgH) 3′ enhancer, elevating MYC expression as a mechanism instigating Burkitt lymphoma.121-124 In multiple myeloma, a t(4:14) translocation involving the IgH locus upregulates FGFR3 and MMSET expression via acquisition of IgH 3′ and intronic enhancers, respectively.125,126 MMSET, which encodes a histone methyltransferase,127 overexpression, but not FGFR3 overexpression, is implicated in myelomagenesis.128,129
Given cancer cell genomic instability, presumably, oncogenic mechanisms involving a chromosomal rearrangement that leads to enhancer expropriation by a gene, resulting in transcriptional induction and a growth and/or survival advantage, are not rare. The chromatin landscape is crucial for deciphering these scenarios, as insulators130 and other elements may negate the actions of surreptitiously introduced enhancers, rendering them inactive or diverting their activities to other genes, while protecting the nearest neighbors.
A scenario analogous to MYC has emerged with the human GATA2 enhancer counterpart (h-77) to the −77 enhancer. Although +9.5 mutations can cause GATA2-deficiency syndrome,51,68 h-77 point mutations have not been reported. In poor-prognosis 3q21;q26 AML, an inversion repositions ∼18 kb of sequence, harboring h-77, ∼4 Mb upstream of GATA2 next to MECOM encoding the leukemogenic protein EVI1. Studies with human cells and mice indicate that h-77–induced EVI1 expression, concomitant with GATA2 loss, constitutes the leukemogenic mechanism.131-133 TALEN-mediated excision of the repositioned h-77 in MUTZ-3 AML cells liberates a maturation blockade, resulting in differentiation into monocyte/macrophage-like cells.132 Because deleting −77 strongly reduces Gata2 expression,48 this has important implications for EVI1 upregulation in AML. Removing h-77 from GATA2 would decrease GATA2 levels, raising the question of which factors drive h-77 activity to increase EVI1 transcription. Other than occupancy by GATA2 and factors that colocalize with GATA2 (eg, LDB1),48 mechanisms underlying −77 (and h-77) activity are unresolved.
Corrupting, creating, and expropriating enhancers: general principles
Genetic and epigenetic aberrations corrupt, create, and expropriate enhancers (Figure 3) to cause, promote, or suppress blood pathologies. The examples described herein highlight the impact of mutations and chromosomal aberrations on enhancer-dependent oncogenic mechanisms involving HSPCs. Epigenetic mechanisms are also critical, although the complexity of the consequences differs greatly from enhancer corruption, which often dysregulates a limited cadre of nearest-neighbor genes. Altered levels or activity of a chromatin regulator, such as a histone methyltransferase, can elicit broad-sweeping epigenome remodeling over a wide swath of the genome. Ascribing the contribution of individual enhancers and genes to cellular phenotypes is extremely complicated. In the case of GATA2 enhancer corruption, because GATA2 regulates a large target gene ensemble, this aberration may derail many cellular processes, secondarily to the primary impact on GATA2 expression.

Corrupting, creating, and expropriating enhancers through mutation or chromosomal aberrations. By destroying cis-element integrity and impairing factor binding, mutations corrupt enhancer function. Corruption may involve a complete loss of activity (if the factor affected is essential), partial loss of activity (if the remaining enhancer components can confer some degree of transcriptional activation), or acquisition of ectopic activity distinct from that of the wild-type enhancer. Mutations may generate cis elements that permit transcription factor binding, following by binding of additional transcription factors and coregulators to generate an enhancer at a site normally lacking an enhancer. Chromosomal inversions or translocations can expropriate a gene’s enhancer, transferring it to another gene, thus creating an ectopic gene-regulatory mechanism. A, gene normally controlled by enhancer; B, gene that expropriates the enhancer.
Corrupting, creating, and expropriating enhancers through mutation or chromosomal aberrations. By destroying cis-element integrity and impairing factor binding, mutations corrupt enhancer function. Corruption may involve a complete loss of activity (if the factor affected is essential), partial loss of activity (if the remaining enhancer components can confer some degree of transcriptional activation), or acquisition of ectopic activity distinct from that of the wild-type enhancer. Mutations may generate cis elements that permit transcription factor binding, following by binding of additional transcription factors and coregulators to generate an enhancer at a site normally lacking an enhancer. Chromosomal inversions or translocations can expropriate a gene’s enhancer, transferring it to another gene, thus creating an ectopic gene-regulatory mechanism. A, gene normally controlled by enhancer; B, gene that expropriates the enhancer.
Considering that multiprotein complexes drive enhancer function, and posttranslational modifications are prevalent in the proteome, altered expression or activity of enzymes mediating these modifications constitutes another mode of dysregulating enhancer function. Signaling mechanisms regulate proteins occupying enhancers and their partners tethered via protein-protein interactions. An instructive example of how oncogenic signaling corrupts a signal-dependent enhancer mechanism emerged from the extensively studied c-Myc oncoprotein. Notch1 signaling activates a long-range MYC enhancer that promotes thymocyte development. Dysregulated Notch1 signaling in T-ALL alters enhancer activity as an oncogenic mechanism.134 At first glance, it would seem that disrupting signal-dependent transcriptional mechanisms affects broad target gene ensembles. However, such mechanisms can exert context-dependent influences on transcription factor function at restricted target gene cohorts. Oncogenic Ras signaling induces GATA2 multisite phosphorylation, which increases its activity at only select target genes.60,135 Although many questions exist regarding the mechanistic basis of context-dependent cellular signaling, this is likely related to differential coregulator requirements for transcription factor function at distinct target genes.136,137 Chromatin access, complex assembly, and coregulator recruitment and utilization all represent steps in which signaling mechanisms differentially influence different loci in distinct subnuclear environments.
A central question relates to why certain enhancers are essential for transcriptional activation, whereas others are modulatory or seem to lack activity. This is crucial when considering how mutations in specific motifs within an enhancer affect function. A single motif within a cluster of motifs may contribute qualitatively or quantitatively to enhancer function or be redundant with other motifs within the cluster. A single-nucleotide change in a motif might abrogate or attenuate factor binding, enhance binding, or impact binding dynamics and therefore complex assembly. Mutations can generate factor-binding sites permissive for factor occupancy in chromatin and enhancer generation. While considering a cancer cell genome rife with mutations and rearrangements, one can envision that these aberrations will create enhancers, while corrupting others, and expropriate enhancers to genes that are not normally enhancer-dependent. If these events occur in a relatively homogenous cell population, for example, a predominant clone in clonal hematopoiesis, it is feasible to deploy current technologies to map prospective enhancers and gene activity and piece together this muddled landscape. However, considering tumor cell heterogeneity and the diversity of genome scrambling in different cells of a tumor, this is a much more daunting problem to contemplate. Much more work is required to determine the impact of genetic variation on enhancer corruption, creation, and expropriation in vivo and devise strategies to mitigate deleterious actions of rogue enhancers operational in contexts in which they should not exist. Mechanistic advances will elevate genome-reading perspicuity and invariably accelerate clinical genetic and precision medicine opportunities.
Acknowledgments
This work was supported by research funding from the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (DK50107 and DK68634).
Authorship
Contribution: E.H.B. wrote the manuscript; and K.D.J. edited the review.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Emery H. Bresnick, Department of Cell and Regenerative Biology, School of Medicine and Public Health, University of Wisconsin–Madison, 4009 WIMR, 1111 Highland Ave, Madison, WI 53705; e-mail: ehbresni@wisc.edu.

