

Key Points
In vivo deficiency of sf3b1 leads to defects in erythroid maturation and proliferation.
TGFβ inhibition releases cell-cycle arrest in sf3b1-mutant erythrocytes, but worsens anemia.

Abstract
The spliceosomal component Splicing Factor 3B, subunit 1 (SF3B1) is one of the most prevalently mutated factors in the bone marrow failure disorder myelodysplastic syndrome. There is a strong clinical correlation between SF3B1 mutations and erythroid defects, such as refractory anemia with ringed sideroblasts, but the role of SF3B1 in normal erythroid development is largely unknown. Loss-of-function zebrafish mutants for sf3b1 develop a macrocytic anemia. Here, we explore the underlying mechanism for anemia associated with sf3b1 deficiency in vivo. We found that sf3b1 mutant erythroid progenitors display a G0/G1 cell-cycle arrest with mutant erythrocytes showing signs of immaturity. RNA-sequencing analysis of sf3b1 mutant erythroid progenitors revealed normal expression of red blood cell regulators such as gata1, globin genes, and heme biosynthetic factors, but upregulation of genes in the transforming growth factor β (TGFβ) pathway. As TGFβ signaling is a known inducer of quiescence, the data suggest that activation of the pathway could trigger sf3b1 deficiency–induced anemia via cell-cycle arrest. Indeed, we found that inhibition of TGFβ signaling released the G0/G1 block in erythroid progenitors. Surprisingly, removal of this checkpoint enhanced rather than suppressed the anemia, indicating that the TGFβ-mediated cell-cycle arrest is protective for sf3b1-mutant erythrocytes. Together, these data suggest that macrocytic anemia arising from Sf3b1 deficiency is likely due to pleiotropic and distinct effects on cell-cycle progression and maturation.
Introduction
The spliceosome is a macromolecular machine with the primary function of removing noncoding introns from pre–messenger RNA (mRNA). Alternative splicing that generates different isoforms of mature mRNA can greatly increase proteome diversity. Systematic RNA sequencing of human erythrocytes from progenitor stages throughout differentiation revealed complex alternative splicing at each step of erythroid maturation.1 This finding suggests that splicing is critical for erythropoiesis, but how splicing factors impact erythroid proliferation and maturation in vivo is largely unknown.
Factors involved with pre-mRNA processing are commonly mutated in human hematopoietic diseases, especially those resulting in anemia and erythropoietic dysplasia.2-4 For example, Splicing Factor 3B, subunit 1 (SF3B1), a core component of the spliceosome, is one of the most prevalently mutated factors in myelodysplastic syndromes (MDS).2-4 Mutations in SF3B1 correlate strongly with a subcategory of MDSs characterized by refractory anemia with ring sideroblasts,5-7 although the exact mechanism that connects genotype to phenotype has not been found. Additionally, SF3B1 itself undergoes extensive alternative splicing during erythroid differentiation, suggesting that SF3B1 could have discrete roles in distinct stages of erythropoiesis.8
Uncovering the in vivo function of SF3B1 in erythropoiesis has been hampered due to the early lethality of Sf3b1-null mice before the formation of the blood system.9 In contrast, zebrafish loss-of-function zygotic mutants are viable until ∼3 days postfertilization (dpf) due to maternally contributed Sf3b1 protein and mRNA.10 This maternal contribution means loss-of-function sf3b1 mutants are functionally similar to a hypomorph, and survive to a developmental time point that allows for characterization of primitive erythroid differentiation. Previously, we demonstrated that zebrafish loss-of-function sf3b1 mutants display a macrocytic anemia characterized by diminished hemoglobinized erythrocytes that have immature and dysplastic morphology.10 Here, we show that the anemia caused by sf3b1 deficiency is due to both maturation defects and a decrease in erythrocyte number. Using RNA sequencing, we identified elevated expression of transforming growth factor β (TGFβ) and p53 pathways and a decrease in the expression of cell-cycle programs in sf3b1-mutant gata1:eGFP+ erythroid progenitors. There were also extensive alterations in splicing with exon-skipping events most prevalent. Through pharmacological and genetic inhibition, we found that TGFβ signaling, but not p53, induced a cell-cycle arrest that protected sf3b1 mutants from a more severe anemia. Our data imply that Sf3b1 regulation of erythropoiesis has pleiotropic and distinct effects on cell-cycle progression and maturation.
Methods
Zebrafish
Zebrafish were maintained as described.11 All fish were maintained according to institutional animal care and use committee–approved protocols in accordance with Albert Einstein College of Medicine research guidelines.
Zebrafish lines
The sf3b1-mutant line hi3394a contains a viral insertion between the first and second exons of the sf3b1 gene, resulting in a premature stop codon in exon 2.12,13 Wild-type and heterozygous embryos are phenotypically indistinguishable from one another, thus, for all of the experiments presented in this manuscript, wild- type and sf3b1-heterozygous embryos are cumulatively referred to as siblings. As sf3b1-homozygous mutants are developmentally delayed and show defects in melanocyte development,14 mutant embryos were age-matched for all analyses according to morphological features as best as possible. Mutant embryos die between 2 and 3 dpf, precluding analysis of later stages.13 For flow cytometry analysis, we generated sf3b1hi3394a/+;Tg(gata1:eGFP)15 animals. For studies examining Tp53, we crossed sf3b1 heterozygous to the well-established tp53zdf1 mutant that has diminished Tp53 activity.16
Whole-mount in situ hybridization and o-dianisidine staining
In situ hybridization for hbbe3,17 cmyb,18 and gata119 was performed as described previously by Thisse et al20 with minor modifications: before proteinase K (Roche) permeabilization, embryos were bleached after rehydration to remove pigmentation. The bleaching was done for 5 to 10 minutes using a bleaching solution of 0.8% KOH, 0.9% H2O2, and 0.1% Tween 20.
O-dianisidine staining was performed as described previously.21 Briefly, dechorionated live embryos were soaked in o-dianisidine staining solution (0.62 mg/mL o-dianisidine [Sigma-Aldrich], 10.9 μM sodium acetate, and 0.65% H2O2) for 10 minutes in the dark, then rinsed in phosphate-buffered solution plus 0.1% Tween 20 (PBT) twice and observed under the microscope. Afterward, embryos were preserved in a 4% paraformaldehyde (PFA) solution.
For in situ hybridization following o-dianisidine staining, we performed the o-dianisidine protocol as described in the previous paragraph until the PBT rinse step; afterward, we followed the in situ hybridization steps as described previously by Thisse et al20 with minor modifications: embryos were transferred to methanol for dehydration directly after o-dianisidine staining without fixing in 4% PFA. Methanol dehydration was done for 2 hours at −20°C.
Flow cytometry
For generation of single-cell suspensions, embryos were first removed from their chorions using pronase (Roche), and then homogenized by manual dissociation using a sterile razor blade followed by digestion with Liberase (Roche). For the digestion, dissociated embryos were resuspended in 1× Dulbecco phosphate-buffered saline (PBS; D-PBS) (Life Technologies) supplemented with a 1/65 dilution of 5 mg/mL Liberase and then incubated at 37°C for 6 minutes. The reaction was stopped with 5% fetal bovine serum (FBS) (Life Technologies). The cells were then filtered once through a 40-μm cell strainer (Falcon) and pelleted by centrifugation at 900g for 5 minutes. Cell pellets were resuspended in fluorescence-activated cell sorting buffer (0.9× D-PBS, 5% FBS, 1% Penn/Strep [Life Technologies]). 4′,6-Diamidino-2-phenylindole (DAPI) was added to a final concentration of 1 μg/mL to identify and exclude dead cells from the analysis. Samples were analyzed with a LSRII flow cytometer (BD Biosciences) and FlowJo version 10.0.8.
Cell-cycle analysis
For cell-cycle analysis, cells were prepared as described in the previous section, except after the first centrifugation, the cell pellets were resuspended in 1 mL of 4% PFA solution (Affymetrix) and incubated at room temperature for 15 minutes. Cells were pelleted again, supernatant was removed, cells were resuspended in D-PBS, and then stained with an anti–green fluorescent protein (GFP) antibody (Life Technologies). Afterward, cells were pelleted, supernatant was removed, and cells were resuspended in a D-PBS solution to which DAPI was added to a final concentration of 1 μg/mL. Samples were analyzed with an LSRII flow cytometer (BD Biosciences) and FlowJo version 10.0.8.
Erythroid cell RNA sequencing
Cell sorting.
Embryos from an incross of sf3b1 Tg(gata1:eGFP) fish were divided into siblings and mutants, and prepared for flow cytometry as described in "Flow Cytometry." For each sample, 30 000 gata1-eGFP+ cells from sf3b1 mutant and siblings were sorted directly into 800 μL of Lysis buffer from the Quick-RNA MicroPrep kit (Zymo Research). Samples were immediately frozen at −80°C until ready to proceed to RNA extraction.
RNA extraction.
We followed the Quick-RNA MicroPrep kit (Zymo Research) protocol, with minor modifications: step 3 was omitted. The DNase-free DNA TURBO kit (Life Technologies) was used for DNA removal at the end of the extraction.
Library preparation.
Quality and concentration of RNA and libraries was determined using the Bioanalyzer RNA Pico Chips, performed by the Albert Einstein College of Medicine Genomics Core facilities. Ribosomal RNA (rRNA) was removed with the RiboGone Mammalian kit (Clontech). Libraries were made following the protocol and using the reagents from the SMARTer Stranded RNA-Seq kit (Clontech).
Computational analysis and pathway analysis.
RNA-sequencing data were generated from rRNA-depleted RNA for 5 sibling RNA samples and 5 mutant RNA samples. Analyses used the following genome release: D.rerio (GRcz10) and the GRCz10.85.gtf annotation file. Reads were trimmed using TRIM GALORE software to check for quality of 150-bp reads. These paired-end reads were mapped using STAR aligner (version 2.4.2a) to the reference genome. Differential expression analysis was done using DESeq222 and EdgeR23 Bioconductor packages. To detect alternative splicing events, we used rMATS24 (version 3.2.5). Reads were trimmed to fixed length of 100 bp and were remapped using STAR aligner before running rMATS. To calculate fragments per kilobase per million reads mapped, Cufflinks (version 2.2.1) was used. Pathway analysis was done with mSigDB.25,26
Treatment with SB431542
Embryos from an incross of sf3b1-heterozygous fish were treated with 25 μM TGFβ inhibitor SB431542 (Selleckchem) in the dark from 7 hours postfertilization (hpf) until 24 hpf for cell-cycle analysis or until 48 hpf for o-dianisidine staining. For the 48-hpf time point, drug was refreshed at 24 hpf. Doses tested were based on a previous study.27 Control embryos were treated with equal volume of dimethyl sulfoxide (DMSO) as a vehicle control.
Morpholino injections
To knockdown tp53 levels, we used a published translation-inhibiting morpholino.28 Embryos from an sf3b1-heterozygous incross were injected with 8 ng of morpholino and compared with noninjected sibling controls.
Statistics
Each experiment was performed with a minimum of triplicate repeats. For pairwise comparisons, the Student t test (unpaired, 2-tailed) was used. Analysis of variance (ANOVA) with a Bonferroni false discovery rate (FDR) multitesting correction was performed when multiple groups were compared in an experiment. For comparison of probabilities, a Fisher's exact test was used.
Results
sf3b1 mutants have fewer, less mature erythrocytes
Anemia can arise for numerous reasons. We had previously determined that the anemia in sf3b1 mutants was associated with defects in erythroid maturation as demonstrated by diminished levels of hemoglobinized cells with more immature morphology compared with their wild-type siblings.10 Diminished erythrocyte number, defects in erythroblast maturation, or hemoglobinization can lead to a decrease in the amount of hemoglobinized cells. Based on the observation that sf3b1-mutant erythroid cells were at various stages of differentiation according to morphology,10 we hypothesized that mutants contained 2 separate pools of erythrocytes: immature, nonhemoglobinized cells and more mature cells that are hemoglobinized. To further explore this hypothesis, we decided to assess the maturation state of erythrocytes using a metric independent of hemoglobinization. Normally, the early embryonic globin gene hbbe3 is nearly absent in wild-type embryos by 48 hpf.17 Indeed, at 36 hpf, wild-type siblings already have diminished levels of hbbe3, with little to no expression by 48 hpf (Figure 1A; supplemental Figure 1). In sf3b1 mutants at 36 and 48 hpf, some embryos had a large amount of hbbe3-expressing cells, whereas others had none (Figure 1B; supplemental Figure 1). Similarly, o-dianisidine staining in 48-hpf mutant embryos also followed the same pattern: in some embryos, there is a large amount of hemoglobinized erythrocytes, whereas in other embryos hemoglobinized erythrocytes are completely absent (Figure 1C-D). This gives rise to 2 possibilities:
There could be 2 developmentally distinct erythrocyte populations in the mutant embryos at 48 hpf: 1 population that is immature and still expressing hbbe3 and not functionally hemoglobinized, and 1 population that matures normally and becomes hemoglobinized; or
There could be a single erythrocyte population in the mutant embryos that is immature but is capable of carrying oxygen by 48 hpf.
To distinguish between both possibilities, we performed sequential o-dianisidine staining followed by hbbe3 in situ hybridization in the same sf3b1-mutant embryos (Figure 1E). Using this approach, we can visualize whether the hbbe3-expressing cells also possess functional hemoglobin within an individual embryo. The results showed that, in sf3b1 mutants, all o-dianisidine–positive cells are also positive for hbbe3 expression (Figure 1F). The failure of mutant erythroid cells to downregulate hbbe3 is strong evidence that mutant red blood cells are not maturing like wild-type cells. Combined, this evidence suggests that the diminishment in hemoglobinized erythrocytes in sf3b1 mutants at 48 hpf results from the mutants having fewer erythrocytes, which are also less mature.
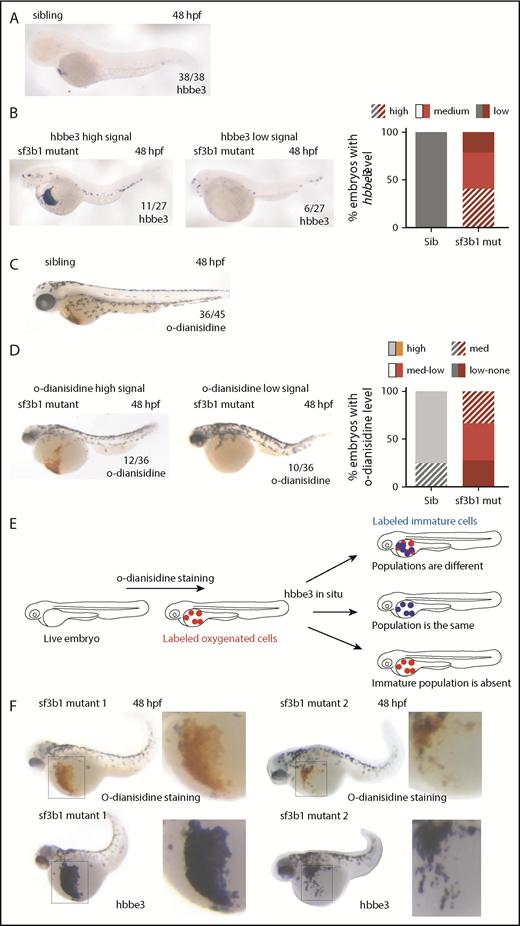
Sf3b1 regulates erythrocyte number and maturation. (A-D) In situ hybridization of early globin hbbe3 (A-B) and o-dianisidine staining (C-D) in siblings (Sib) (A,C) and sf3b1 mutant (mut) (B,D) at 48 hpf. For mutants, example of a high signal embryo is shown on the left and an example of a mutant with low signal is shown on the right. Numbers in the lower right denote number of embryos that displayed a similar phenotype to the image. Graphs on the right denote the percentage of embryos with the designated phenotype. (E) Schematic representing the experimental flow to measure hbbe3 levels in o-dianisidine–positive erythrocytes by sequential staining. O-dianisidine staining was performed first followed by hbbe3 in situ hybridization in the same embryo, thus consecutively labeling oxygenated and immature erythrocytes. (F) O-dianisidine staining (top) followed by in situ hybridization of hbbe3 (bottom) in the same sf3b1-mutant embryos at 48 hpf (2 examples shown). Inset to the right shows a higher magnification view of area boxed in the image on the left. (A-D,F) In situ stain nitroblue tetrazolium, o-dianisidine stain; original magnification ×6, inset ×12. Med, medium.
Sf3b1 regulates erythrocyte number and maturation. (A-D) In situ hybridization of early globin hbbe3 (A-B) and o-dianisidine staining (C-D) in siblings (Sib) (A,C) and sf3b1 mutant (mut) (B,D) at 48 hpf. For mutants, example of a high signal embryo is shown on the left and an example of a mutant with low signal is shown on the right. Numbers in the lower right denote number of embryos that displayed a similar phenotype to the image. Graphs on the right denote the percentage of embryos with the designated phenotype. (E) Schematic representing the experimental flow to measure hbbe3 levels in o-dianisidine–positive erythrocytes by sequential staining. O-dianisidine staining was performed first followed by hbbe3 in situ hybridization in the same embryo, thus consecutively labeling oxygenated and immature erythrocytes. (F) O-dianisidine staining (top) followed by in situ hybridization of hbbe3 (bottom) in the same sf3b1-mutant embryos at 48 hpf (2 examples shown). Inset to the right shows a higher magnification view of area boxed in the image on the left. (A-D,F) In situ stain nitroblue tetrazolium, o-dianisidine stain; original magnification ×6, inset ×12. Med, medium.
Erythroid progenitors arising from both primitive and erythroid-myeloid progenitor (EMP)-derived definitive waves are present during the developmental time points analyzed in Figure 1. To determine whether there were defects in EMP-derived erythropoiesis, we performed in situ hybridization for the progenitor marker c-myb at 30 and 36 hpf and gata1 at 30 hpf in siblings and sf3b1 mutants (supplemental Figure 2A-B). Expression of both c-myb and gata1 within the posterior blood island (PBI) region where EMPs form was similar between siblings and sf3b1 mutants. Furthermore, we examined erythroid progenitor levels at 24 and 36 hpf within the PBI using Tg(gata1:eGFP) zebrafish,15 in which enhanced GFP (eGFP) marks erythroid cells, and observed high levels of gata1:eGFP within the PBI region of mutant embryos (supplemental Figure 2C). In a prior study,10 we examined the expression of a myeloid-associated EMP marker (pu.1) and also found normal expression in sf3b1 mutants. Combined these data indicate that EMP formation is unaffected when levels of sf3b1 are limiting. Mutants become progressively unhealthier from 48 to 72 hpf, thus we were not able to assess differentiation of EMP-derived erythrocytes. Based on these data, we conclude that the majority of the erythrocyte defects observed up to 48 hpf in sf3b1 mutants are due to defects in primitive wave-derived cells.
Cell-cycle arrest is triggered in sf3b1-mutant erythrocytes
To quantify erythrocyte numbers at 24, 36, and 48 hpf, we measured the frequency and absolute number of gata1:eGFP+ cells in siblings and sf3b1 mutants using flow cytometry (Figure 2A-C). No difference was observed between siblings and mutants at 24 hpf. Both frequency and absolute number of gata1:eGFP+ erythrocytes were diminished at 36 hpf compared with siblings with the effect even more pronounced by 48 hpf.
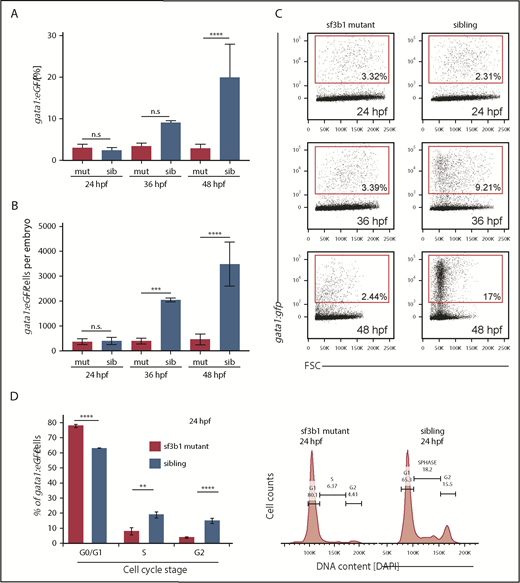
Loss of sf3b1 triggers a G0/G1 cell-cycle arrest in erythroid progenitors. (A-B) Graph quantifying gata1:eGFP+ cells in sf3b1-mutant and wild-type siblings at 24, 36, and 48 hpf. (A) Percentage of gata1:eGFP+ cells. (B) Absolute number of gata1:eGFP+ cells per embryos. (C) Representative flow cytometry plots for panels A and B. (D) Graph quantifying the percentage of gata1:eGFP+ erythrocytes in G0/G1, S, or G2/M phases of the cell cycle at 24 hpf. Representative flow cytometry histograms of erythrocyte DNA content as measured by DAPI fluorescence intensity on sf3b1 mutants and siblings. All experiments were done in biological triplicates. Statistical significance calculated by an ANOVA with a Bonferroni FDR multitesting correction. **P < .01, ***P < .001, ****P < .0001. FSC, forward scatter; n.s., not significant.
Loss of sf3b1 triggers a G0/G1 cell-cycle arrest in erythroid progenitors. (A-B) Graph quantifying gata1:eGFP+ cells in sf3b1-mutant and wild-type siblings at 24, 36, and 48 hpf. (A) Percentage of gata1:eGFP+ cells. (B) Absolute number of gata1:eGFP+ cells per embryos. (C) Representative flow cytometry plots for panels A and B. (D) Graph quantifying the percentage of gata1:eGFP+ erythrocytes in G0/G1, S, or G2/M phases of the cell cycle at 24 hpf. Representative flow cytometry histograms of erythrocyte DNA content as measured by DAPI fluorescence intensity on sf3b1 mutants and siblings. All experiments were done in biological triplicates. Statistical significance calculated by an ANOVA with a Bonferroni FDR multitesting correction. **P < .01, ***P < .001, ****P < .0001. FSC, forward scatter; n.s., not significant.
These findings suggest a lack of erythrocyte expansion or elevated cell death. From our previous work, we observed no increase in active Caspase-3 staining in the blood-forming regions of sf3b1 mutants compared with siblings, suggesting apoptosis is not driving the lower erythrocyte numbers in mutants.10 Next, we tested whether differences in proliferation could underlie the decrease in erythrocyte frequency. Cell-cycle status of gata1:eGFP+ erythroid progenitors from siblings and sf3b1 mutants at 24 hpf was quantified via flow cytometry using DAPI fluorescence as a metric for DNA content (Figure 2D). Mutant cells had an increased amount of cells in the G0/G1 phase (2N content of DNA) and a greatly diminished amount of cells in S phase in comparison with their wild-type siblings. These results suggest that sf3b1 loss in erythrocytes triggers a G0/G1 cell-cycle arrest.
Expression and splicing of the TGFβ and p53 pathways are altered in sf3b1-mutant erythrocytes
To understand the mechanism behind the erythroid defect in sf3b1 mutants, we conducted RNA sequencing of purified erythroid progenitors from wild-type and mutant siblings at 24 hpf when differences in erythroid cell number are not yet evident in the mutant embryos. This permitted us to capture some of the earliest transcriptional changes leading to erythrocyte dysfunction. We sorted gata1:eGFP+ cells from siblings and sf3b1 mutants at 24 hpf. Stranded RNA-sequencing libraries were then generated from rRNA-depleted RNA samples. Five independent biological replicates were analyzed. On average, ∼28 million paired-end 150-bp sequencing reads were acquired per sample. DE-Seq222 and EdgeR23 were used to determine differential gene expression (defined as twofold change, FDR P < .05) between control sibling and mutant erythrocytes. Of the nearly 32 000 genes analyzed, 913 genes were downregulated and 774 were upregulated in sf3b1 mutants compared with sibling control erythrocytes (Figure 3A-B; supplemental Tables 1 and 2). The heatmap shows the top 500 differentially expressed genes and clustering of the 5 replicates according to genotype (Figure 3C). Among the normally expressed genes are several erythropoiesis regulators, all embryonic globins, and most of the heme-group synthesis pathway genes (Figure 3D).
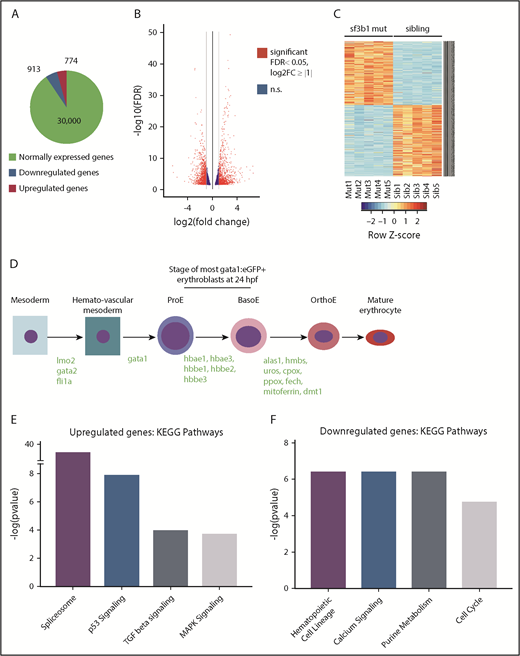
TGFβ- and Tp53-regulated genes are upregulated in sf3b1-mutant erythroid progenitors. (A) Representative chart of gene expression in gata1:eGFP+ cells from sf3b1 mutants. (B) Volcano plot displaying differentially expressed genes between gata1:eGFP+ cells from sf3b1 mutants and siblings. Significant differences are defined as FDR <0.05 and log2 fold change (FC) >1. Gray lines denote the fold-change threshold. (C) Heatmap of top 500 differentially expressed genes between gata1:eGFP+ cells from sf3b1 mutants and siblings. Each row is a gene and each column is a replicate. Mutant samples are shown on the left and sibling samples are shown on the right. (D) Schematic highlighting normally expressed hematopoietic genes in sf3b1-mutant erythrocytes and the stage at which they begin to be expressed (gene names in green). (E-F) Representative charts of significantly altered KEGG pathways in genes upregulated (E) or downregulated (F) in sf3b1-mutant erythroid progenitors compared with sibling controls as determined by mSigDB analysis. BasoE, basophilic erythroblast; OrthoE, orthochromatophilic erythroblast; ProE, proerythroblast.
TGFβ- and Tp53-regulated genes are upregulated in sf3b1-mutant erythroid progenitors. (A) Representative chart of gene expression in gata1:eGFP+ cells from sf3b1 mutants. (B) Volcano plot displaying differentially expressed genes between gata1:eGFP+ cells from sf3b1 mutants and siblings. Significant differences are defined as FDR <0.05 and log2 fold change (FC) >1. Gray lines denote the fold-change threshold. (C) Heatmap of top 500 differentially expressed genes between gata1:eGFP+ cells from sf3b1 mutants and siblings. Each row is a gene and each column is a replicate. Mutant samples are shown on the left and sibling samples are shown on the right. (D) Schematic highlighting normally expressed hematopoietic genes in sf3b1-mutant erythrocytes and the stage at which they begin to be expressed (gene names in green). (E-F) Representative charts of significantly altered KEGG pathways in genes upregulated (E) or downregulated (F) in sf3b1-mutant erythroid progenitors compared with sibling controls as determined by mSigDB analysis. BasoE, basophilic erythroblast; OrthoE, orthochromatophilic erythroblast; ProE, proerythroblast.
To gain insight into the affected pathways, the upregulated and downregulated gene lists were analyzed with mSigDB, a platform that computes overlaps between experimentally derived gene lists and lists of genes in known pathways.25,26 Specifically, we compared the upregulated and downregulated gene sets in sf3b1-mutant erythroid progenitors to the Kyoto Encyclopedia of Genes and Genomes (KEGG) collection, which contains 186 gene sets collated for biological function.29-31 In the upregulated gene list, spliceosome was the top enriched gene set suggesting a compensatory upregulation of splicing factors due to loss of sf3b1 (Figure 3E; supplemental Table 3). Additionally, genes involved with the stress-activated p53-signaling pathway, TGFβ-signaling pathway, and MAPK were also among the top enriched pathways in the upregulated gene set. Of note, activation of both the p53 and TGFβ pathways is linked with cell-cycle arrest and cellular quiescence in hematopoietic cells.32,33 In the downregulated list, genes associated with hematopoietic cell lineage were among the top enriched gene set, consistent with the deregulation of erythroid maturation in sf3b1 mutants (Figure 3F; supplemental Table 4). In line with the observed cell-cycle arrest, factors involved with the cell cycle were also significantly enriched in the downregulated genes. Together, these data show that sf3b1-mutant erythrocytes have deregulated expression of genes involved with hematopoietic lineage, cell cycle, and signaling.
Sf3b1 is a part of the U2 small nuclear ribonucleoprotein complex important for pre-mRNA splicing, thus we also examined how sf3b1 loss affected mRNA splicing in embryonic erythrocytes. Differential inclusion rates across 16 766 splicing events between sf3b1 mutants and wild-type siblings were calculated using replicate Multivariate Analysis of Transcript Splicing (rMATS).24 Splicing events with inclusion differences ≥10% and FDR ≤0.01 were considered significantly misspliced. A total of 1357 events in 1104 genes were misspliced between sf3b1 mutants and siblings with cassette exons being the largest group (Figure 4A; supplemental Table 5). The volcano plots show splicing events with a change in the percent spliced in (Δψ) between control siblings (<0) and sf3b1 mutants (>0) (Figure 4B). In 82% of the misspliced transcripts, only a single differential event was detected indicating a high degree of specificity for the splicing events that are most sensitive to Sf3b1 levels. Aberrant splicing can result in the inclusion of premature nonsense codons that then trigger transcript degradation via the nonsense-mediated decay pathway.34 We compared the misspliced genes to those differentially expressed between sf3b1 mutants and siblings and found no significant overlap suggesting the alterations in splicing are not majorly contributing to changes in overall transcript abundance in erythroid progenitors (Figure 4C).
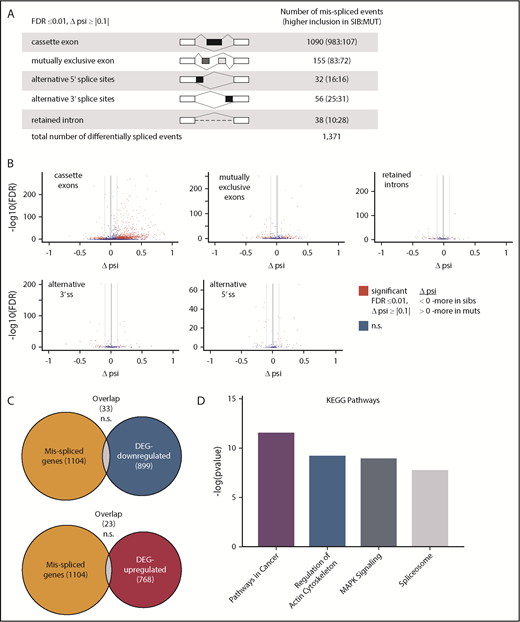
Cancer-associated genes are misspliced in sf3b1-mutant erythroid progenitors. (A) Schematic of the different splicing events detected by analysis with rMATS. The numbers in the parentheses (n1:n2) indicate the number of significant events that have higher inclusion level for sibling (n1) or for mutant (n2). (B) Volcano plot of differential splicing events between gata1:eGFP+ cells from sf3b1 mutants and siblings. Significant differences are defined as FDR ≤0.01 and Δψ > |0.1|. Gray lines denote the Δψ threshold. (C) Venn diagrams showing overlap of misspliced transcripts with differentially expressed genes. Probability of overlap calculated by hypergeometric distribution. (D) Representative charts of significantly altered KEGG pathways in misspliced transcripts in sf3b1-mutant erythrocytes compared with sibling controls as determined by mSigDB analysis. DEG, differentially expressed genes; psi, percent spliced in; ss, splice site.
Cancer-associated genes are misspliced in sf3b1-mutant erythroid progenitors. (A) Schematic of the different splicing events detected by analysis with rMATS. The numbers in the parentheses (n1:n2) indicate the number of significant events that have higher inclusion level for sibling (n1) or for mutant (n2). (B) Volcano plot of differential splicing events between gata1:eGFP+ cells from sf3b1 mutants and siblings. Significant differences are defined as FDR ≤0.01 and Δψ > |0.1|. Gray lines denote the Δψ threshold. (C) Venn diagrams showing overlap of misspliced transcripts with differentially expressed genes. Probability of overlap calculated by hypergeometric distribution. (D) Representative charts of significantly altered KEGG pathways in misspliced transcripts in sf3b1-mutant erythrocytes compared with sibling controls as determined by mSigDB analysis. DEG, differentially expressed genes; psi, percent spliced in; ss, splice site.
Altered splicing can also change the function of the protein encoded by the transcript. To determine whether particular pathways were impacted in the misspliced genes, we compared the misspliced gene list to the KEGG gene sets using mSigDB. Some of the top pathways enriched in the misspliced genes were associated with cancer (pathways in cancer, MAPK signaling, and spliceosome) (Figure 4D; supplemental Table 6). Although the overlap of the misspliced genes and differentially expressed genes was moderate, similar pathways were affected, suggesting functional convergence of altered splicing and gene expression when sf3b1 is mutated.
Sf3b1 regulation of TGFβ is essential for erythrocyte maturation and expansion
The gene-expression data suggest that activation of the TGFβ or p53 pathways could contribute to the anemia in sf3b1 mutants. TGFβ is a versatile pathway that plays several different roles in cellular regulation.35 TGFβ signaling is linked to erythroid differentiation and is elevated in MDS; inhibitors of TGFβ superfamily signaling are currently being tested clinically for treatment of a variety of ineffective erythropoiesis syndromes, including MDS-associated refractory anemia.36-38 These findings combined with the observation that genes in the TGFβ-signaling pathway are upregulated in sf3b1-mutant erythroid progenitors led us to test the effect of inhibiting TGFβ signaling on the erythropoietic defects in sf3b1 mutants.
To test this hypothesis, we treated embryos with the TGFβ inhibitor SB431542, previously shown to inhibit Smad3-dependent TGFβ signaling in zebrafish, and then determined the effects on cell-cycle progression and maturation (Figure 5A).39 TGFβ signaling can suppress erythroid progenitor proliferation,40 thus we determined whether the cell-cycle arrest in sf3b1-mutant erythrocytes was due to TGFβ signaling. Cell-cycle status of 24-hpf sf3b1-mutant gata1:eGFP+ erythroid progenitors was assessed in embryos treated with 25 μM SB431542 or DMSO control (Figure 5A). There was a significant increase of gata1:eGFP+ cells in S phase and concomitant decrease of cells in G0/G1 (Figure 5B). Next, we examined the impact on erythroid maturation at 48 hpf and found the surprising result that sf3b1 mutants treated with 25 μM SB431542 displayed a more severe anemia than those treated with the DMSO control (Figure 5C). Together, these data indicate that TGFβ signaling triggers a G0/G1 cell-cycle arrest in sf3b1-mutant erythrocytes that protects the mutant cells from later maturation defects and/or cell death.
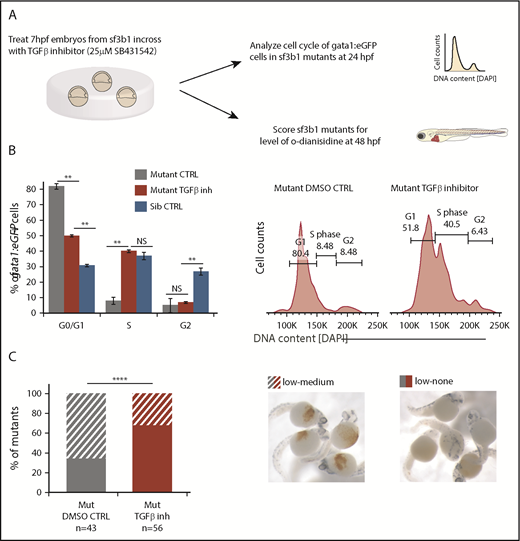
TGFβ-mediated cell-cycle arrest is protective for sf3b1-mutant erythrocytes. (A) Schematic of experiment to determine whether inhibition of TGFβ signaling alters erythroid cell-cycle arrest or anemia in sf3b1 mutants. (B) Graph quantifying the percentage of gata1:eGFP+ erythrocytes in G0/G1, S, or G2/M phases of the cell cycle at 24 hpf in sf3b1 mutants treated with a DMSO control or 25 μM of the TGFβ inhibitor SB431542. Cell cycle of wild-type siblings is included for comparison. Representative flow cytometry histograms of erythrocyte DNA content as measured by DAPI fluorescence intensity on sf3b1 mutants treated with a DMSO control or 25 μM SB431542 are shown on the right. Statistical significance calculated by an ANOVA with a Bonferroni FDR multitesting correction. (C) Graph showing frequency of sf3b1 mutants with designated levels of o-dianisidine–positive oxygenated erythrocytes at 48 hpf after treatment with DMSO control or 25 μM SB431542. Total number of mutants analyzed per treatment group is listed below the graph. Images of sf3b1-mutant embryos with low-medium or low-none o-dianisidine–positive cells are shown to the right. Significance of comparison between groups determined by the Fisher's exact test. All experiments were done in biological triplicates. **P < .01, ****P < .0001. (C) O-dianisidine stain; original magnification ×4.
TGFβ-mediated cell-cycle arrest is protective for sf3b1-mutant erythrocytes. (A) Schematic of experiment to determine whether inhibition of TGFβ signaling alters erythroid cell-cycle arrest or anemia in sf3b1 mutants. (B) Graph quantifying the percentage of gata1:eGFP+ erythrocytes in G0/G1, S, or G2/M phases of the cell cycle at 24 hpf in sf3b1 mutants treated with a DMSO control or 25 μM of the TGFβ inhibitor SB431542. Cell cycle of wild-type siblings is included for comparison. Representative flow cytometry histograms of erythrocyte DNA content as measured by DAPI fluorescence intensity on sf3b1 mutants treated with a DMSO control or 25 μM SB431542 are shown on the right. Statistical significance calculated by an ANOVA with a Bonferroni FDR multitesting correction. (C) Graph showing frequency of sf3b1 mutants with designated levels of o-dianisidine–positive oxygenated erythrocytes at 48 hpf after treatment with DMSO control or 25 μM SB431542. Total number of mutants analyzed per treatment group is listed below the graph. Images of sf3b1-mutant embryos with low-medium or low-none o-dianisidine–positive cells are shown to the right. Significance of comparison between groups determined by the Fisher's exact test. All experiments were done in biological triplicates. **P < .01, ****P < .0001. (C) O-dianisidine stain; original magnification ×4.
Dampening p53 signaling can improve anemia in some zebrafish mutants including those with mutations in ribosomal components.28 Tp53 is a potent inducer of cell-cycle arrest, thus we examined whether knockdown of tp53 using a well-established morpholino28 could further improve cell-cycle progression in sf3b1 mutants treated with the TGFβ inhibitor (supplemental Figure 3A-B). We observed no further improvement in cell-cycle progression upon tp53 knockdown compared with TGFβ inhibition alone. Next, we determined whether diminished Tp53 activity affects erythroid maturation by measuring o-dianisidine levels in sf3b1;tp53zdf1 double mutants, which have diminished Tp53 activity.16 The frequency of sf3b1 mutants with medium-low o-dianisidine-positive erythrocytes was decreased in sf3b1;tp53 double mutants compared with single mutants (supplemental Figure 3B). The same result was seen in sf3b1 mutants injected with the tp53 morpholino (data not shown). Double sf3b1;tp53 mutants treated with the TGFβ inhibitor showed the same effect as those only treated with the TGFβ inhibitor.
Discussion
This study of erythroid cell development in sf3b1-mutant embryos has uncovered important details about the effects of spliceosomal disruption on red blood cell biology. We found that sf3b1 deficiency led to the production of fewer erythrocytes that show maturation defects and dysplasia.10 Genes in the TGFβ and the p53 pathways were significantly upregulated upon sf3b1 loss. Functionally, TGFβ pathway activity induced a G0/G1 cell-cycle arrest in sf3b1-mutant proerythroblasts. Pharmacological inhibition of the TGFβ pathway relieved the cell-cycle arrest, but worsened anemia. These data suggest that loss of sf3b1 triggers a TGFβ-mediated cell-cycle checkpoint that acts to protect erythroid progenitors, possibly from aberrant differentiation due to diminished amounts of Sf3b1. Additionally, our findings suggest that TGFβ signaling plays a more prominent role in sf3b1-mutant anemia than Tp53. Combined, these data demonstrate a key functional difference between anemia caused by loss of sf3b1 vs those with ribosomal defects.
Few studies have explored the function of wild-type SF3B1 in erythropoiesis. A recent report demonstrated that in vitro knockdown of SF3B1 in human CD34+ hematopoietic stem and progenitor cells results in ineffective erythropoiesis.41 In this study, activation of the p53 pathway contributed to cell-cycle arrest and elevated apoptosis in early-stage progenitors. In zebrafish, we similarly found that the p53 pathway was activated, but, unlike in the in vitro study, this had little effect on erythropoiesis in sf3b1-mutant cells, indicating key functional differences in response to splicing factor deficiency in cell culture vs an animal model. There is growing evidence that the microenvironment is heavily involved in MDS and ineffective erythropoiesis.42 Indeed, our findings in an sf3b1-deficient zebrafish model support a role for the microenvironment in anemia highlighting the importance of animal models in understanding how splicing factors regulate hematopoiesis.
Unlike the zebrafish sf3b1 mutants that are homozygous loss of function, MDS-associated mutations in SF3B1 are mainly heterozygous missense mutations.7 The most common mutations are in the C-terminal portion of the protein most often changing lysine 700 to glutamic acid (K700E). Murine models with hematopoietic-restricted expression of Sf3b1+/K700E develop macrocytic anemia and have a G0/G1 arrest in early progenitors similar to zebrafish sf3b1-homozygous mutants.43,44 The prevailing view of how MDS-associated mutations like K700E affect SF3B1 is that they change SF3B1 splice-site preferences and result in dominant neomorphic functions in splicing.43-45 Despite these differences in splicing function, over 10% of genes misspliced in SF3B1-mutated MDS46 were also misspliced in zebrafish sf3b1-mutant erythrocytes. At the expression level, 37% of KEGG pathways enriched in the deregulated genes in SF3B1-mutated MDS CD34+ progenitors compared with healthy controls47 were also enriched in the genes differentially expressed in zebrafish sf3b1-mutant erythrocytes. Although the zebrafish sf3b1 loss-of-function mutants show more extensive transcriptome changes, these cellular and molecular commonalities suggest that there is some similarity between zebrafish sf3b1 mutants and SF3B1-mutated MDS.
Clinically, novel TGFβ superfamily inhibitors are currently being tested for treatment of low-risk MDS, which are highly enriched for SF3B1-mutated MDS.36,37 These protein-based drugs, such as sotatercept and luspatercept, work as activin ligand traps that prevent receptor-mediated activation of the pathway. These TGFβ superfamily inhibitors seem to work by inhibiting the growth differentiation factor 11 (GDF11) ligand, which results in stimulation to erythroid differentiation in late-stage erythropoiesis, distinct from the action of erythropoietin that stimulates early-stage progenitors. In zebrafish sf3b1 mutants, treatment with TGFβ receptor kinase inhibitor SB431542 resulted in improvements in erythroid progenitor proliferation, but a worsening of anemia. Our study demonstrates that inhibition of type I TGFβ receptors have the potential to improve cell-cycle progression of arrested erythroid progenitors, but cannot improve differentiation. Future studies are warranted to examine the impact of dual treatment with both types of TGFβ inhibitors.
SF3B1-modulating drugs are also being evaluated for the treatment of patients with splicing factor-mutated MDS.48 These drugs work via a synthetic lethal modality that leads to the selective death of splicing factor mutant cells over nonmutated cells. Our findings suggest that treatment with a TGFβ receptor inhibitor plus an SF3B1 modulator could enhance synthetic lethality to ablate splicing factor mutant clones. So far, TGFβ superfamily inhibitors show minimal long-term side effects, whereas splicing factor modulating drugs can have adverse side effects.49 Combined treatment could result in a more robust effect at lower drug doses resulting in safer treatment.
Our study provides mechanistic insight into how Sf3b1 defects drive aberrant erythropoiesis in vivo. The findings highlight the importance of animal models to understand how defects in this highly relevant MDS-associated factor impacts erythropoiesis.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Amit Verma, Eirini Trompouki, and Charles Query for helpful discussions on this work. The authors acknowledge the assistance of numerous core facilities at Albert Einstein College of Medicine, including Flow Cytometry, Genomics, and Epigenomics Facilities and the Zebrafish Core Facility. The graphical abstract was created with biorender.com.
This work was supported by the Gabrielle’s Angel Foundation, the American Cancer Society (RSG-129527-DDC), the Kimmel Foundation, and the EvansMDS Foundation (T.V.B.). V.G. was supported by National Institues of Health, National Institute of General Medical Sciences grant GM57829. The Albert Einstein College of Medicine Flow Cytometry, Genomics, and Epigenomics Facilities were supported by National Institutes of Health, National Cancer Institute cancer grant P30CA013330.
Authorship
Contribution: A.D.L.G., R.C.C., E.F., S.N., and T.V.B. performed experiments and analyzed data; V.G. performed bioinformatics analyses; and A.D.L.G. and T.V.B. wrote and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for A.D.L.G. is Biogen, Durham, NC.
The current affiliation for R.C.C. is Ferrier Research Institute, Victoria University of Wellington, Wellington, New Zealand.
Correspondence: Teresa V. Bowman, Albert Einstein College of Medicine, 1300 Morris Park Ave, Chanin 501, Bronx, NY 10461; e-mail: teresa.bowman@einstein.yu.edu.

