

Key Points
Reduction of APL antibodies by immunoadsorption may be a lifesaving therapy for the management of DAH with high titer of APL antibodies.
Autologous HSCT may be a valid treatment option in patients with primary APS and no response to standard immunosuppressive therapy.
Introduction
The antiphospholipid syndrome (APS) is a rare systemic autoimmune disease defined by thrombotic events or pregnancy morbidity occurring in patients with persistent antiphospholipid (APL) antibodies.1
Although thrombosis is the most common cause of pulmonary injury, there are also other lung conditions affecting these patients, including diffuse alveolar hemorrhage (DAH).2-9 This latter manifestation suggests that APL antibodies can promote tissue lesions by other means than thrombosis.
We hypothesized that immunoadsorption would rapidly decrease circulating APL antibodies and save the life of a patient presenting with recurrent severe DAH. Envisioning long-term outcomes, we further assumed that autologous hematopoietic stem cell transplantation (AHSCT) would reset the immune system, and thereby reduce the production of APL antibodies preventing further episodes of DAH and succeed where all immunosuppressive treatment options had failed.
Case description
A 48-year-old man developed progressive dyspnea over the course of 3 to 4 weeks, as well as livedo reticularis on his right foot. Seven years ago, he was diagnosed with a triple-positive (ie, positive for lupus anticoagulant, anticardiolipin antibodies, and anti-β2-glycoprotein 1 antibodies) primary APS after an unprovoked pulmonary embolism. The patient has also a past history of myocardial infarction and mitral valve disease, both related to the diagnosis of APS.
A contrast-enhanced thoracic-computer tomography showed bilateral diffuse ground-glass lesions, suggestive of alveolar hemorrhages (Figure 1A), without signs of pulmonary embolism or signs of active bronchial artery bleeding. A bronchoscopy confirmed the presence of blood within both lungs. Microbiologic tests and cultures for bacteria, fungi, viruses, and mycobacteria, as well as search for neoplastic cells from bronchoalveolar lavage, were negative. Cytological examination showed numerous macrophages intensely stained by Prussian blue, denoting iron, reflecting severe intra-alveolar hemorrhage (Figure 1B-C). Lupus anticoagulant was positive, and anticardiolipin and anti-β2-glycoprotein 1 immunoglobulin G antibody titers were very high (Figure 2). Notably, the rest of the immune-serological work-up showed that antinuclear antibodies were only transiently positive (1:320). However, anti-centromere proteins, anti-double stranded DNA, anti-nucleosomes, anti-histones, anti-RPN, anti-PCNA, anti-ribosomes, anti-SS-A and SS-B, anti-Scl-70, anti-RNA polymerase III, anti-Jo-1, anti-Mi-2, anti-Ku, anti-PM-Scl 100, anti-PR3-ANCA, anti-MPO-ANCA, and anti-glomerular basement membrane (Goodpasture) antibodies were negative. These results confirmed the diagnosis of primary APS. IV methylprednisolone boluses followed by oral prednisone were administered (Figure 2). To rapidly reduce APL titers, immunoadsorption on protein A column was conducted (5 courses during a 10-day period, each processing the patient’s plasma volume at least twice). Dyspnea improved promptly after the first immunoadsorption course. To enhance immunosuppression, rituximab was administered (Figure 2). On the eleventh day, the patient was discharged on enoxaparin (80 mg twice daily) and oral prednisone (80 mg/d). Prednisone was slowly tapered and withdrawn after 3 months.
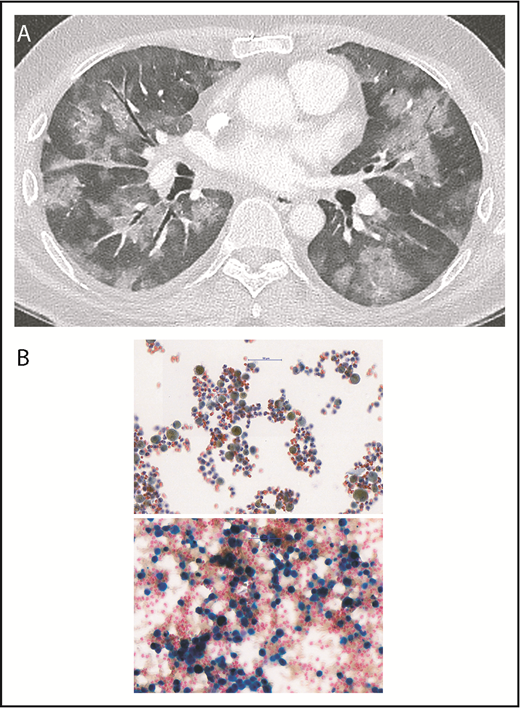
Diffuse alveolar hemorrhage. (A) The contrast-enhanced thoracic-computer tomography shows widespread patchy ground glass opacification and, in part, a faint “crazy paving” pattern. The contrast between the ground glass areas caused by diffuse alveolar hemorrhage and the air containing bronchi render the bronchi more prominent (dark bronchus sign). There were no signs of pulmonary embolism nor signs of active bronchial artery bleeding. (B) Images of cytological bronchoalveolar lavage specimens. (Upper) Routine Papanicolaou staining shows macrophages (50%-60%) and a significant number of neutrophils, as well as lymphocytes. Almost all macrophages contain a brown-yellow pigment in the cytoplasm. (Lower) Prussian blue staining shows an area with more prominent macrophages, of which most stain intensely blue, denoting iron, reflecting severe intra-alveolar hemorrhage (∼95% of the total macrophage population were hemosiderin-laden macrophages, Golde score 250). Magnification, 40×.
Diffuse alveolar hemorrhage. (A) The contrast-enhanced thoracic-computer tomography shows widespread patchy ground glass opacification and, in part, a faint “crazy paving” pattern. The contrast between the ground glass areas caused by diffuse alveolar hemorrhage and the air containing bronchi render the bronchi more prominent (dark bronchus sign). There were no signs of pulmonary embolism nor signs of active bronchial artery bleeding. (B) Images of cytological bronchoalveolar lavage specimens. (Upper) Routine Papanicolaou staining shows macrophages (50%-60%) and a significant number of neutrophils, as well as lymphocytes. Almost all macrophages contain a brown-yellow pigment in the cytoplasm. (Lower) Prussian blue staining shows an area with more prominent macrophages, of which most stain intensely blue, denoting iron, reflecting severe intra-alveolar hemorrhage (∼95% of the total macrophage population were hemosiderin-laden macrophages, Golde score 250). Magnification, 40×.
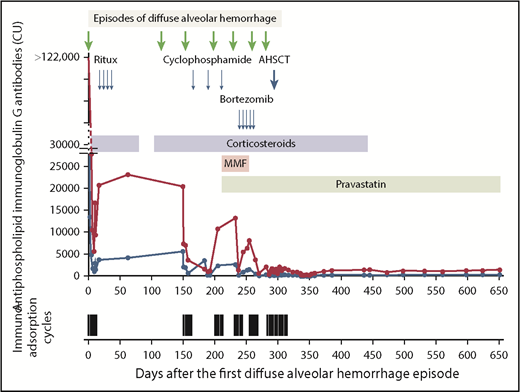
Major events in the clinical course and laboratory results. Shown are the antiphospholipid immunoglobulin G antibody titers (blue line: anticardiolipin; red line: anti-β2-glycoprotein 1) and apheresis cycles. The arrows indicate events and points of intervention. The variation in the anticardiolipin and anti-β2-glycoprotein 1 immunoglobulin G antibody titers until day 315 reflects management with immunoadsorption. Dosages of administered therapy are as follow. Corticosteroids: day 0 to day 79: methylprednisolone boluses (500 mg daily IV) for 3 days followed by prednisone 1 mg/kg daily; day 101 to day 444: methylprednisolone boluses (1000 mg daily IV) for 3 days, followed by prednisone 1 mg/kg per day tapering and withdraw. Rituximab: 375 mg/m2 weekly for 4 weeks. Cyclophosphamide: pulses 750 mg/m2 every 3 weeks. Mycophenolate mofetil: 1 g daily for 1 week, followed by 1 g twice daily. Pravastatin: 40 mg daily. Bortezomib: 1.3 mg/m2 every 72 hours × 5 doses. Peripheral blood stem cell mobilization: cyclophosphamide: 2 g/m2 given IV and granulocyte colony-stimulating factor (filgrastim) at 10 µg per kilogram of body weight administered subcutaneously daily beginning 3 days later. Peripheral blood stem cell collection was performed 10 days after the initiation of the mobilization. The time interval between the apheresis collection of peripheral blood stem cells and the start of the conditioning regimen was 3 days. Conditioning regimen: From day −5 to day −2 (ie, from 5 days to 2 days before transplantation), the patient received a conditioning regimen consisting of cyclophosphamide 50 mg per kilogram of body weight per day given IV on days −5, −4, −3, and −2 (total dose, 200 mg/kg) and equine anti-thymocyte globulin (ATGAM Pfizer) 30 mg per kilogram of body weight given IV on days −4, −3, and −2 (total dose, 90 mg/kg body weight). Immunoadsorption: a total of 12 treatment cycles (4-9 treatments/cycle) were performed. The duration of each treatment was 225-300 minutes. CU, chemiluminescent units; MMF, mycophenolate mofetil; Ritux, rituximab.
Major events in the clinical course and laboratory results. Shown are the antiphospholipid immunoglobulin G antibody titers (blue line: anticardiolipin; red line: anti-β2-glycoprotein 1) and apheresis cycles. The arrows indicate events and points of intervention. The variation in the anticardiolipin and anti-β2-glycoprotein 1 immunoglobulin G antibody titers until day 315 reflects management with immunoadsorption. Dosages of administered therapy are as follow. Corticosteroids: day 0 to day 79: methylprednisolone boluses (500 mg daily IV) for 3 days followed by prednisone 1 mg/kg daily; day 101 to day 444: methylprednisolone boluses (1000 mg daily IV) for 3 days, followed by prednisone 1 mg/kg per day tapering and withdraw. Rituximab: 375 mg/m2 weekly for 4 weeks. Cyclophosphamide: pulses 750 mg/m2 every 3 weeks. Mycophenolate mofetil: 1 g daily for 1 week, followed by 1 g twice daily. Pravastatin: 40 mg daily. Bortezomib: 1.3 mg/m2 every 72 hours × 5 doses. Peripheral blood stem cell mobilization: cyclophosphamide: 2 g/m2 given IV and granulocyte colony-stimulating factor (filgrastim) at 10 µg per kilogram of body weight administered subcutaneously daily beginning 3 days later. Peripheral blood stem cell collection was performed 10 days after the initiation of the mobilization. The time interval between the apheresis collection of peripheral blood stem cells and the start of the conditioning regimen was 3 days. Conditioning regimen: From day −5 to day −2 (ie, from 5 days to 2 days before transplantation), the patient received a conditioning regimen consisting of cyclophosphamide 50 mg per kilogram of body weight per day given IV on days −5, −4, −3, and −2 (total dose, 200 mg/kg) and equine anti-thymocyte globulin (ATGAM Pfizer) 30 mg per kilogram of body weight given IV on days −4, −3, and −2 (total dose, 90 mg/kg body weight). Immunoadsorption: a total of 12 treatment cycles (4-9 treatments/cycle) were performed. The duration of each treatment was 225-300 minutes. CU, chemiluminescent units; MMF, mycophenolate mofetil; Ritux, rituximab.
Four months after discharge, a second episode of DAH ensued. The patient received methylprednisolone boluses for 3 days, followed by oral prednisone. After an initial improvement, the patient developed a new episode of DAH and 5 courses of immunoadsorption were conducted. The patient was discharged 9 days later in good clinical condition with enoxaparin (80 mg twice daily), prednisone therapy (75 mg/d), hydroxychloroquine (200 mg twice daily), and pulsed cyclophosphamide therapy (Figure 2). A fourth episode of DAH occurred 1 day after the second pulse of cyclophosphamide, and a fifth episode 1 day before the fourth scheduled cyclophosphamide pulse. Patient management is described in Figure 2. Cyclophosphamide therapy was interrupted, mycophenolate mofetil and pravastatin were introduced, and the patient was scheduled for 5 courses of bortezomib to target plasma cells. Under treatment with bortezomib, a sixth episode of DAH ensued. Four days after hospital discharge, a seventh episode of DAH occurred, and this time, the patient condition was critical and tracheal intubation was required.
Given the severity and refractoriness to all administered immunosuppressive therapies, AHSCT was performed (Figure 2). Peripheral blood progenitor cell collection was followed by CD34+ cell enrichment of peripheral blood stem cell apheresis product, using paramagnetic nanobead-coupled CD34 antibody and immunomagnetic extraction (CliniMACS Plus, Miltenyi). A total of 290 days after the first episode of DAH (Figure 2), the patient received the autologous peripheral blood stem-cell transplant (3.8 × 106 CD34+ cells/kg). To prevent the recurrence of DAH during the early posttransplant period, immunoadsorption was performed every 2 to 3 days. Prednisone was finally stopped on day 446 after the first episode of DAH, and 156 days after transplantation. At the most recent visit, on day 650 after the first episode of DAH and 1 year after transplantation, the patient was completely asymptomatic and off immunosuppression for more than 200 days. Lupus anticoagulant was undetectable. However, anticardiolipin and anti-β2-glycoprotein 1 immunoglobulin G antibodies were still present, but ∼100-fold lower than at presentation. The CD4+ T-cell count was below the normal range (335 cells per microliter; normal range, 410-1590 cells per microliter), and the regulatory T-cell population was not completely reconstituted (CD4+/CD25+/FOXP3+: 4.9% or 16.4 cells per microliter [normal range, 17.3-26.7 cells per microliter]10 ; CD4+/CD25+/CD127−: 4.2% or 14.07 cells per microliter [normal range, 44.4-68.6 cells per microliter]10 ).
Methods
The report was a retrospective chart review of 1 patient with primary APS and life-threatening DAH. The patient provided written consent for the review of his medical records and publication of the results, per the Declaration of Helsinki.
Immunoadsorption treatments were performed using adsorber columns (Immunosoprba, Fresenius). Plasma was separated using Spectra Optia (Terumo BCT) and a plasma monitor (ADAsorb, Medicap).
Results and discussion
Pulmonary capillaritis is a form of vasculitis that may be limited to the lungs and may cause DAH.11 In our case, DAH was probably caused by pulmonary capillaritis in the setting of primary APS with very high APL antibody titers, as previously described.2-9 There was no evidence of acute thromboembolism or other autoimmune etiologies of DAH. Less than 1% of patients with APS develop DAH.12-14 Affected patients are more often males and are more likely to have high-titer APL antibodies, cardiac valve disease,12-14 livedo reticularis, pulmonary hypertension, stroke, seizures, and pregnancy morbidity.6,8 Our patient is male, had a previous history of cardiac valve disease, and displayed livedo reticularis lesions at the time of the first episode of DAH.
The treatment of DAH has been studied in randomized trials only in the context of severe presentations of granulomatosis with polyangiitis, mixed cryoglobulinemia, and systemic lupus erythematosus. Thus, treatment decisions in other forms are based on extrapolation from these studies, from case series, and from clinical experience.9,15 Most of the diseases causing capillaritis are treated with a combination of systemic corticosteroids and immunosuppressive therapy, such as cyclophosphamide, rituximab, or plasma exchange.15,16 Additional treatment of APL with DAH consists of IV immunoglobulin, hydroxychloroquine, and mycophenolate mofetil.2,15,16 Several studies suggest that statins reduce endothelial activation induced by APL antibodies.17,18 Therefore, we added pravastatin as an adjunct therapy.
Because refractoriness to standard treatment is associated with high risk and poor outcome, the management of these patients is always a challenge.
Rapid elimination of autoantibodies using selective immunoadsorption instead of unselective plasma exchange is a pathophysiologically guided therapeutic approach.19 Because our patient presented with an unusual high titer of APL antibodies, we hypothesized that immunoadsorption would, at least transiently, rapidly and efficiently reduce APL antibodies, offering a lifesaving therapy. The major clinical improvement observed immediately after the first immunoadsorption course in our patient underlined the pathogenic effect of the APL antibodies and supported the use of this selective apheresis method. We did not observe severe adverse events during immunoadsorption. After each cycle (4-9 treatments), we infused immunoglobulins (Privigen, CSL Behring) as a replacement therapy.
This patient responded quickly to high-dose corticosteroids associated with immunoadsorption, but did not display a sustainable response to any of the additional successive immunosuppressive treatments (Figure 2).
A conditioning regimen combining cyclophosphamide and anti-thymocyte globuline followed by AHSCT has been used successfully in patients with APS with refractory systemic lupus erythematosus,20 but was not tested in patients with refractory primary APS. This therapeutic approach was chosen for our patient on the premise of removal of autoreactive immunologic memory cells and to effectively reset the immune system by renewing the CD4+ T cell compartment, especially the regulatory T-cell population. Despite the lack of randomized trials, ex vivo selection of CD34+ hematopoietic stem cells from the graft is a frequently used approach in patients with autoimmune diseases.21
The course in this patient’s severe recurrent DAH suggests that rapid intravascular reduction of APL antibodies concentration by immunoadsorption might constitute a valuable lifesaving therapeutic approach for the emergency management of DAH with high titer of APL antibodies. AHSCT may be a valid treatment option to consider in patients with primary APS and no response to standard immunosuppressive therapy.
Acknowledgments
The authors thank their colleagues at the Inselspital (Bern University Hospital) for collaboration on the care of the patient.
Authorship
Contribution: A.A.-S. and A.R. wrote the manuscript; A.A.-S., A.P., Y.B., and A.R. prepared the figures; and B.M.T., F.F., G.M.B., T.P., A.P., Y.B., T.G., and J.A.K.H. reviewed and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Anne Angelillo-Scherrer, Department of Hematology and Central Hematology Laboratory, Inselspital, Bern University Hospital, University of Bern, CH-3010 Bern, Switzerland; e-mail: anne.angelillo-scherrer@insel.ch.

