

Key Points
Leukemic blasts of a female carrier of an ATRX germline mutation have persistently skewed inactivation of the X chromosome.
Germline mutation in leukemia needs to be interpreted with caution because it is not always pathologic.
Introduction
X-linked α-thalassemia mental retardation syndrome (ATR-X) is a rare familial disorder associated with distinctive craniofacial abnormalities, developmental delay, and α-thalassemia, which predominantly affects males.1-3 Gibbons et al4 first identified the causative gene ATRX on chromosome Xq13.3, which encodes for a chromatin remodeling factor of the SW1/SNF family and modulates transcription regulation, DNA repair, and mitotic recombination.5,6 Recently, somatic mutations of ATRX were reported in various human cancers, including acute lymphoblastic leukemia (ALL).7,8 ATRX deficiency results in genetic instability through impairment of nonhomologous end joining and increase in sensitivity to DNA damage in a mouse glioma model.9 However, the exact role of ATRX in human carcinogenesis remains unknown. Here we present a female carrier of ATR-X who developed highly aggressive pre-B ALL, who relapsed after allogeneic stem cell transplantation. We performed whole-genome sequencing (WGS) and RNA sequencing (RNA-seq) to investigate the role of germline ATRX mutation in the leukemogenesis of this patient.
Case description
A 29-year-old woman was diagnosed with pre-B ALL after presenting with back pain and lymphadenopathy. Her 2 brothers have ATR-X with germline mutations in the ATRX gene (c.4617_4622del; p.1540_1541delDE, NM_000489.3), and she was found to be a carrier. Her mother, the presumed ATRX mutation carrier, was diagnosed with metastatic melanoma, and her maternal uncle died as a result of glioma.
Despite the fact that her first induction therapy failed, she achieved a complete remission with clofarabine-based salvage chemotherapy. Eighteen months after the diagnosis, she developed an extramedullary and central nervous system relapse. She was treated with radiation therapy, blinatumomab, and intrathecal chemotherapy and was referred for allogeneic stem cell transplantation with her father as a donor. The pretransplant evaluation showed a positron emission tomography–avid lesion in the spine and bone marrow involvement of 25% pre-B ALL with a complex cytogenetic abnormality. She received cranial radiation and clofarabine immediately before a myeloablative conditioning regimen using total body irradiation of 12 Gy, fludarabine, donor lymphocyte infusion of 2 × 108 CD3+ cells per kg (day −6), and cyclophosphamide after the donor lymphocyte infusion. A haploidentical peripheral blood stem cell graft of 8.0 × 106 CD34+ cells per kg was infused followed by sirolimus with ultra-low-dose interleukin-2 for graft-versus-host disease prophylaxis. Engraftment was rapid with full donor chimerism of CD3 and myeloid cells by day 14. She developed steroid-responsive grade 2 acute graft-versus-host disease. On day 74, her leukemia relapsed in the bone marrow, which contained 87% leukemic blasts with increased complexities of cytogenetics consistent with clonal evolution. Despite salvage chemotherapy, she died as a result of septic shock and multiorgan failure.
Methods
Biospecimen collection and cell and nucleotide isolation
Informed consent was obtained for research use of biospecimens in compliance with the Declaration of Helsinki under the protocols approved by the Institutional Review Board of the National Heart, Lung, and Blood Institute (ClinicalTrials.gov identifiers NCT02226861 and NCT00071045). Leukemic blasts were flow-sorted on a BD FACSAria II (BD Biosciences, San Jose, CA), and peripheral blood CD3+ T cells (germline control) and donor B cells were isolated using Robosep (STEMCELL Technologies, Vancouver, BC, Canada). Genomic DNA and total RNA were extracted by using a column purification method (Allprep DNA/RNA mini kit, Catalog No. 80204, QIAGEN, Hilden, Germany).
WGS, RNA-seq, and bioinformatic analysis
WGS was performed on 3 samples to cover 100× in 2 leukemia samples (pre- and posttransplant leukemias) and 30× in control cells (germline T cells). RNA-seq with 150 bp and 40 million reads per sample was performed on 4 samples (germline T cells, 2 leukemias, and B cells from the patient’s father). Library generation, reverse transcription, and complementary DNA amplification were performed according to manufacturer’s instructions, and both WGS and RNA-seq libraries were sequenced on Illumina HiSeq 3000 (Illumina, San Diego, CA). Somatic mutations were called by Varscan and Mutect2, followed by annotation using SnpEff. SciClone was used to infer an evolution of subclones.10 Circros graphic software was used for graphic demonstration of the genomic landscape.11 The transcriptome was analyzed using STAR alignment and DESeq.12 Gene Ontology annotation analysis13 and Ingenuity Pathway Analysis (QIAGEN) were used for gene expression pathway analysis (Sequence Read Archive [SRA] submission number, SUB6133095; Bioproject Submission ID, SUB6183192; BioProject ID, PRJNA560392).
Results and discussion
WGS confirmed germline mutation of ATRX (c.4617_4622del; p.1540_1541delDE, NM_000489.3) in both leukemic blasts and T cell controls as a high-confidence germline mutation. However, transcripts of mutant ATRX gene were not detected with RNA-seq in leukemic blasts and normal cells, indicating an inactivation of the mutant X chromosome. Most female carriers of ATR-X are known to have skewed X-chromosome inactivation (XCI) of the mutant ATRX allele. To confirm the selective inactivation of the maternally inherited X chromosome, we analyzed the heterozygosity of multiple single nucleotide polymorphism (SNP) sites on the X chromosome. As shown in Figure 1, most SNPs on the X chromosome (from Xp22.11 to Xq28) were selectively activated in the paternal alleles. Transcriptions of the maternal SNP allele were detectable only in pseudoautosomal region 1 (PAR1) (supplemental Figure 1). This highly skewed inactivation of the maternal X chromosome persisted in leukemic blasts. Expression levels of the entire ATRX transcript by RNA-seq also showed no significant changes in expression between leukemic blasts and controls (supplemental Table 1).

Heatmap of SNP allele expression pattern in X chromosome. Variant allele frequencies (VAFs) of the SNP genotype from WGS data were plotted on the first left column. VAFs of SNP expressions of 4 samples from RNA-seq data were plotted on the right 4 columns. Despite WGS data that show the presence of heterozygosity (ie, VAF ranges from 25% to 75%) in each SNP loci, RNA-seq data demonstrated the loss of heterozygosity (either 0% or 100%) in the majority of leukemia and germline cells, which exclusively expressed SNP alleles inherited from the patient’s father except FAM104B and the pseudoautosomal region (supplemental Figure 1). This finding is consistent with persistent XCI in hematopoietic lineage cells in the patient. Tx, transplant
Heatmap of SNP allele expression pattern in X chromosome. Variant allele frequencies (VAFs) of the SNP genotype from WGS data were plotted on the first left column. VAFs of SNP expressions of 4 samples from RNA-seq data were plotted on the right 4 columns. Despite WGS data that show the presence of heterozygosity (ie, VAF ranges from 25% to 75%) in each SNP loci, RNA-seq data demonstrated the loss of heterozygosity (either 0% or 100%) in the majority of leukemia and germline cells, which exclusively expressed SNP alleles inherited from the patient’s father except FAM104B and the pseudoautosomal region (supplemental Figure 1). This finding is consistent with persistent XCI in hematopoietic lineage cells in the patient. Tx, transplant
WGS identified various somatic mutations, gene fusions (supplemental Table 2), and copy number variation in pretransplant leukemia (Figure 2A) and posttransplant leukemia (Figure 2B). Among somatic mutations, 119 mutations in protein coding genes were persistently detected in both pre- and postleukemic blasts. Clonal inference and evolution analysis using SciClone revealed founding clones (cluster 1) with high variant allele frequency and 3 additional subclones. One of 3 subclones (cluster 2) showed an increase in variant allele frequency after transplantation, which suggests that the clones inherited survival advantages, possibly escaping from graft-versus-leukemia effects (supplemental Figure 2; supplemental Table 3). RNA-seq analysis revealed upregulation of genes in the NOD-like receptor and JAK-STAT signaling pathway in posttransplant blasts compared with pretransplant blasts (supplemental Figures 3 and 4).
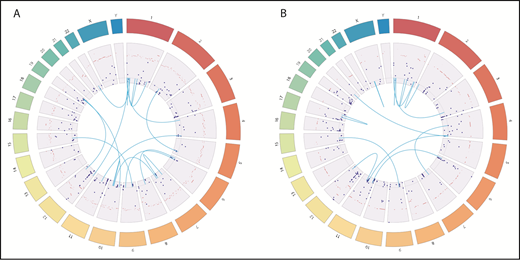
Genomic landscape of somatic mutations, gene fusions, and copy number variations in leukemia. Dark blue dots represent somatic mutation with VAFs. Light blue lines represent fusion genes. Dark bars represent copy number variations. (A) In pretransplant leukemia, WGS and RNA-seq identified 270 somatic mutations (251 mutations in protein-coding genes, 16 in intergenic regions, and 3 in noncoding RNA), 16 fusion genes, and 725 gene loci with copy number variations. (B) Posttransplant leukemia harbors 312 somatic mutations (296 in protein coding genes, 19 in intergenic regions, and 6 in noncoding RNA), 13 fusion genes, and 332 gene loci with copy number variations.
Genomic landscape of somatic mutations, gene fusions, and copy number variations in leukemia. Dark blue dots represent somatic mutation with VAFs. Light blue lines represent fusion genes. Dark bars represent copy number variations. (A) In pretransplant leukemia, WGS and RNA-seq identified 270 somatic mutations (251 mutations in protein-coding genes, 16 in intergenic regions, and 3 in noncoding RNA), 16 fusion genes, and 725 gene loci with copy number variations. (B) Posttransplant leukemia harbors 312 somatic mutations (296 in protein coding genes, 19 in intergenic regions, and 6 in noncoding RNA), 13 fusion genes, and 332 gene loci with copy number variations.
Here we report a female carrier of an ATRX germline mutation with a strong family history of malignancy who developed a highly aggressive pre-B ALL. Notably, the leukemia could not be attributed to the ATRX mutation because the leukemic blasts consistently retained an inactive X chromosome bearing the mutant ATRX. Instead, the leukemia cells demonstrated numerous non-ATRX mutations, which may explain pathophysiology of this highly aggressive and refractory leukemia.
Recently, 2 independent groups14,15 reported osteosarcomas in male ATR-X patients, suggesting genetic predisposition of germline ATRX mutations to cancer development. In contrast, ATRX germline mutation was not a provocative event for the B-ALL in our heterozygous female carrier. In a mouse model, skewed XCI in female carriers occurred over time by selection of the cells expressing the wild-type ATRX allele. The hematopoietic cells underwent selection at the earliest stage of definitive hematopoiesis, resulting in highly skewed XCI in adult blood.16 Therefore, it is likely that XCI in hematopoietic lineage cells is highly conserved, even in leukemic blasts. However, ATRX mutations may play a different role in nonhematologic cancers. For example, ATRX is one of the genes involved in escape from X-inactivation tumor suppressors contributing to cancer sex bias.17 ATRX mutations have been associated with low-grade gliomas in males, whereas escape from X inactivation of ATRX has been described in female brain tissue. Our case study indicates that the biological significance of germline ATRX mutation can vary according to sex and tumor type and that germline ATRX mutation needs to be interpreted with caution because it is not always pathologic in tumor cells.
The full-text version of this article contains a data supplement.
Authorship
Contribution: C.P.B., A.J.B., R.J.G., and S.I. designed and conceived the study; C.P.B., R.P., and S.I. collected the data from the in vitro experiment; C.P.B., C.C., K.A.O., K.R.C., C. Yan, A.J.B., R.J.G., and S.I. analyzed and interpreted the data; C.P.B., C.C., K.A.O., N.D.M., M.B., A.J.B., R.J.G., and S.I. drafted the manuscript; C.C. and C. Yan performed the bioinformatic and statistical analyses; S.H., K.A.O., C. Yuan, P.A.P., N.D.M., M.C.F., M.B., A.J.B., R.J.G., and S.I. collected the clinical data; and A.J.B. and S.I. obtained funding and supervised the study.
Conflict-of-interest disclosure: N.D.M. received research and travel funding from Ventana Biosystems and collaborated on research with ArcherDx. M.C.F. received an honorarium from Shire Plc and a grant from Bellicum Pharmaceuticals, and his institution received fees from Novartis Pharmaceuticals and Amgen Inc. P.A.P. received honoraria from DAVA Pharmaceuticals, served in a consulting or advisory role for France Foundation, and was a member of the speaker’s bureau for Celgene. The remaining authors declare no competing financial interests.
The current affiliation for M.B. is Sarah Cannon Research Institute, Nashville, TN.
The current affiliation for A.J.B. is George Washington Cancer Center, George Washington University, Washington, DC.
Correspondence: Sawa Ito, University of Pittsburgh, UPMC Hillman Cancer Center, 5115 Centre Ave, Pittsburgh, PA 15213; e-mail: itos3@upmc.edu.

