

Abstract
Sickle cell disease (SCD) is an inherited hemoglobinopathy caused by a single point mutation in the β-globin gene. As a consequence, deoxygenated hemoglobin polymerizes triggering red blood cell sickling and hemolysis, vaso-occlusion, and ischemia/reperfusion. Allied to these pathologies is the overproduction of reactive oxygen species driven by hemoglobin Fenton chemistry and peroxidase reactions as well as by secondary activation of vascular oxidases, including NAD(P)H oxidase and xanthine oxidase. In addition, hypoxia, produced by sickle red blood cell occlusion, disrupts mitochondrial metabolism and generates excess superoxide through electron leak from the mitochondrial respiratory chain. Superoxide dismutase 2 (SOD2) is a mitochondrial-specific antioxidant enzyme that dismutates superoxide to hydrogen peroxide, which is then converted to water by catalase and glutathione peroxidase. In SCD, the antioxidant defense system is significantly diminished through decreased expression and activity levels of antioxidant enzymes, including superoxide dismutase, catalase, and glutathione peroxidase. From a translational perspective, genetic variants including a missense variant in SOD2 (valine to alanine at position 16) are present in 45% of people with African ancestry and are associated with increased sickle complications. While it is known that there is an imbalance between oxidative species and antioxidant defenses in SCD, much more investigation is warranted. This review summarizes our current understanding of antioxidant defense systems in SCD, particularly focused on SOD2, and provides insight into challenges and opportunities as the field moves forward.
Introduction
Sickle cell disease (SCD) is the consequence of a single nucleotide substitution (GTG for GAG) at the sixth amino acid of the β-globin gene, which is located on the short arm of chromosome 11.1 This nucleotide change substitutes nonpolar, hydrophobic valine for negatively charged glutamic acid on the surface of the β-globin chain, leading to the polymerization of deoxygenated, abnormal sickle hemoglobin (HbS). HbS polymerization deforms the red blood cell (RBC) into a sickle, crescent-like shape. Repeated sickling of RBCs results in membrane fragility and hemolysis, occlusion of postcapillary venules, ischemia-reperfusion, and infarction. These pathological events augment the generation of free radicals through activation of pro-oxidant enzymes, disruption of mitochondrial respiratory chain activity,2 hemolysis-induced release of free hemoglobin and heme that catalyze the Fenton reaction, and RBC autooxidation.3-8 Thus, the overproduction of free radicals contributes to increased oxidative stress in endothelial cells, RBCs, neutrophils, and platelets, which manifests as multiorgan vasculopathy.3-8
Oxidative stress is defined as the imbalance between reactive oxygen species (ROS), reactive nitrogen species (RNS), and antioxidants. ROS are the reduced metabolites of molecular oxygen. These products are superoxide anion radical (O2⋅−), hydrogen peroxide (H2O2), hydroxyl anion (OH−), and hydroxyl radical (⋅OH).9,10 RNS are the overproduction of nitric oxide (⋅NO) and nitrogen dioxide (⋅NO2) or peroxynitrite (ONOO−), which is formed from the interaction between NO and superoxide. To combat ROS and RNS, both nonenzymatic and enzymatic defense mechanisms have evolved.3 The enzymatic antioxidants include superoxide dismutase (SOD), catalase, glutathione peroxidase, and peroxiredoxins.3 The nonenzymatic antioxidants include carotenoids, ascorbic acid, zinc, and riboflavin.3 Thus, if an imbalance between reactive species is not neutralized by these defense mechanisms, oxidation of biological molecules such as DNA, lipids, proteins, and carbohydrates persists, leading to cell dysfunction and death.3
Despite high oxidative stress, levels of both nonenzymatic11 and enzymatic antioxidants are reduced in SCD.12,13 A wide range of nonenzymatic antioxidants have been found to be deficient in RBCs, platelets, and mononuclear cells of SCD patients, including vitamins E and C, β-carotene, ω-3 fatty acids, and plasma retinol.11,14 Serum and plasma levels of the enzymatic antioxidants SOD, catalase, and glutathione peroxidase are also diminished.12-14 The reduction of the antioxidant defense system has been associated with increased oxidative damage, as measured by plasma lipid and RBC membrane peroxidation, DNA damage markers, and protein carbonylation.12,14 Therapeutic treatments with antioxidants have shown beneficial effects in both in vitro15-17 and in vivo16,18 models of SCD. Validating the preclinical studies, the only 2 US Food and Drug Administration–approved drugs for the treatment of SCD, hydroxyurea and l-glutamine, have antioxidant activity. Hydroxyurea inhibits DNA synthesis, and by complex mechanisms including epigenetic modifications, posttranscriptional, and signaling pathways, it increases levels of fetal hemoglobin. As a result, hydroxyurea reduces the occurrence of vaso-occlusive crises in SCD.19 SCD patients taking hydroxyurea have increased antioxidant capacity and decreased thiobarbituric acid reactive substances (TBARSs), a marker of lipid peroxidation, in their plasma.20 l-glutamine was recently approved by the US Food and Drug Administration and reduces pain crises and the number of hospitalizations due to SCD-related complications.21 The mechanism of action of l-glutamine is currently not fully understood but is presumed to reduce oxidative damage in RBCs.22 Glutamine supplementation is also able to improve antioxidant capacity through regeneration of glutathione.23 The improvement in pathology with antioxidant supplementation underscores the importance of maintenance of the antioxidant defense system in the management of SCD.
SOD2
SODs are a family of enzymatic antioxidant proteins that include 3 isoforms, all with similar activities, that have been identified and described in mammals: copper-zinc SOD (SOD1), manganese SOD (SOD2), and extracellular SOD (SOD3). These enzymes specialize in eliminating oxidative stress through the rapid (catalytic efficiency of 109 M−1 s−1) dismutation of superoxide anion radicals (O2⋅− + O2⋅− + 2H+ → O2 + H2O2)24,25 (Figure 1). Hydrogen peroxide is then further catalyzed to water by catalase, peroxiredoxins, or glutathione peroxidases. The 3 isoforms differ in chromosomal position, gene encoding, protein structure, cellular localization, and cofactor requirements (reviewed in detail in Miao and St Clair25 ). SOD2 is transcribed from sod2 and translated as an ∼23-kDa homotetramer symmetrically assembled from pairs of N-terminal helical hairpins. The helical hairpins along with the C-terminal α/β domains contribute ligands to the formation of the catalytic manganese site within each subunit.26 SOD2 is synthesized in the cytoplasm and localizes to the mitochondrial matrix, where it scavenges and accelerates the dismutation of superoxide anion generated by respiratory chain enzymes.27 During normal erythropoiesis, RBCs lose their mitochondria and other organelles as they mature. This process may be impaired in SCD. A study by Jagadeeswaran et al using reticulocyte-identifying fluorescence-conjugated CD71 and the RNA dye thiazole orange showed increased levels of mitochondria in RBCs.28 This finding was interpreted as increased retention of mitochondria in sickle RBCs, resulting from disrupted hematopoiesis.28 Increased ROS production from RBC mitochondria was also observed.28 It is possible that what was observed in the study by Jagadeeswaran et al was not a retention of mitochondria but a stimulation of erythropoiesis by the severe hemolytic anemia resulting in an increase in the production of many young erythroid precursors that retain membrane-bound organelles such as mitochondria.
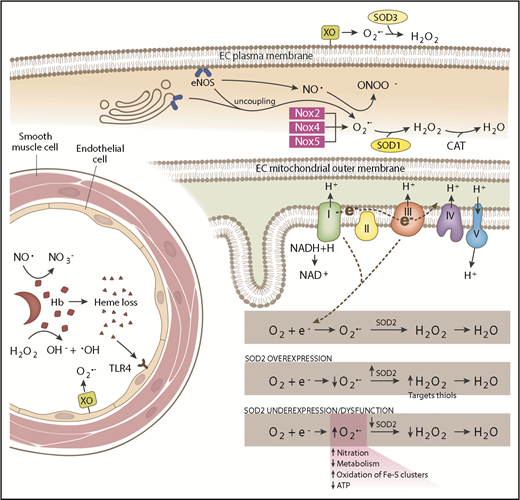
Schematic of SCD-associated oxidative stress pathways and antioxidant systems. In the intravascular space, sickled RBCs undergo hemolysis, releasing hemoglobin (Hb) and heme. Hemoglobin reacts with (1) NO·, forming nitrate (NO3−), and (2) hydrogen peroxide (H2O2), producing hydroxyl radical (−OH). Endothelial-bound xanthine oxidase (XO) generates superoxide (O2·−) and H2O2. Free heme binds to Toll-like receptor 4 (TLR4) producing reactive species through the activation of the NFκB pathway. SOD3 is found in the extracellular compartment and converts O2·− to H2O2. In the endothelial cell (EC) cytoplasm, endothelial NO synthase (eNOS) uncoupling and NADPH oxidase 2 (Nox2), Nox4, and Nox5 produce O2·−, which is dismutated by SOD1. eNOS normally generates NO·, which is capable of reacting with O2·− to form peroxynitrite (ONOO−). In the mitochondrial matrix, electrons leaked from complexes I and III of the respiratory chain react with oxygen (O2), forming O2·−. SOD2 dismutates O2·− to H2O2, which is further broken down to water by catalase (CAT). SOD2 expression and activity is maintained at a precise balance. Overexpression of SOD2 decreases O2·− levels and increases H2O2 levels, which are then free to oxidize protein thiols. SOD2 underexpression or dysfunction increases O2·− levels, increasing nitration and oxidation of iron sulfur clusters and decreasing metabolism and adenosine triphosphate (ATP) production.
Schematic of SCD-associated oxidative stress pathways and antioxidant systems. In the intravascular space, sickled RBCs undergo hemolysis, releasing hemoglobin (Hb) and heme. Hemoglobin reacts with (1) NO·, forming nitrate (NO3−), and (2) hydrogen peroxide (H2O2), producing hydroxyl radical (−OH). Endothelial-bound xanthine oxidase (XO) generates superoxide (O2·−) and H2O2. Free heme binds to Toll-like receptor 4 (TLR4) producing reactive species through the activation of the NFκB pathway. SOD3 is found in the extracellular compartment and converts O2·− to H2O2. In the endothelial cell (EC) cytoplasm, endothelial NO synthase (eNOS) uncoupling and NADPH oxidase 2 (Nox2), Nox4, and Nox5 produce O2·−, which is dismutated by SOD1. eNOS normally generates NO·, which is capable of reacting with O2·− to form peroxynitrite (ONOO−). In the mitochondrial matrix, electrons leaked from complexes I and III of the respiratory chain react with oxygen (O2), forming O2·−. SOD2 dismutates O2·− to H2O2, which is further broken down to water by catalase (CAT). SOD2 expression and activity is maintained at a precise balance. Overexpression of SOD2 decreases O2·− levels and increases H2O2 levels, which are then free to oxidize protein thiols. SOD2 underexpression or dysfunction increases O2·− levels, increasing nitration and oxidation of iron sulfur clusters and decreasing metabolism and adenosine triphosphate (ATP) production.
SOD2 regulation
Given the importance of SOD2 in modulating mitochondrial ROS, an understanding of how it is regulated, particularly in the context of increased oxidative states, can lead to better insights regarding where therapeutic intervention may be beneficial. The human sod2 gene is located on the q25.3 band of chromosome 6 and contains many sites for transcriptional modification: GC-rich motifs, a Foxoa3 binding site, a NF-κB regulatory element, and several specific protein 1 and activator protein 2 sites.25 Transcriptional activation of early growth response 1 (Egr-1) upregulates messenger RNA expression of SOD2 through the induced expression of platelet-derived growth factor.29 Additionally, a variety of proinflammatory cytokines, such as interleukin 1 (IL-1), IL-4, IL-6, tumor necrosis factor α, interferon γ, and the bacterial endotoxin lipopolysaccharide, are robust SOD2 activators.30 In particular, IL-1 and tumor necrosis factor α transcriptionally upregulate expression of manganese SOD (SOD2) in rat pulmonary endothelial cells, a response that was exacerbated by the addition of lipopolysaccharide.31 Developmental regulation of SOD2 was found in mouse kidneys and is associated with mpv17-like protein. Expression levels of both SOD2 and mpv17-like protein increase during embryogenesis, peaking in adulthood before decreasing with age.32,33 An incidental side effect of microtubule formation inhibitor anticancer drugs (vinblastine, Taxol, and vincristine) is transcriptional upregulation of SOD2 through activation of protein kinase C, shown in a human lung adenocarcinoma cell line (A549).34 Downregulation of SOD2 is elicited by the activator protein 2 family transcription factors.35,36 Enzymatic activity of SOD2 is regulated posttranslationally, with phosphorylation at serine 106 by Cdk1 increasing enzymatic activity and cell survival,37,38 while loss of SIRT3 leads to acetylation of lysine 122 and lysine 68 and inhibition of enzymatic activity.39,40
SOD2 expression is also regulated by ROS and RNS. Increased peroxynitrite can cause nitration at tyrosine 34 leading to enzymatic inhibition,41 while superoxide radicals upregulate SOD2 expression through activation of the redox-sensitive transcription factors NF-κB and Nrf2.42,43 The antagonistic roles that peroxynitrite and superoxide radicals have in regulating SOD2 expression and activity aides in understanding why mitochondrial antioxidant response in SCD is particularly dysregulated. In addition to ROS and RNS having opposed regulatory functions, SOD2 activation by superoxide radicals in SCD is impaired. While NF-κB is activated in SCD,44,45 there is diminished Nrf2 activation in transgenic mouse models of SCD, suggesting a dysfunctional response by the sickle antioxidant system. Pharmacological activation of the Nrf2 pathway by treatment with dimethylfumarate (DMF) in transgenic sickle mice is able to increase antioxidant expression (hemoxygenase-1, haptoglobin, hemopexin, and ferritin heavy chain) in the liver and kidneys, as well as prevent progression of disease pathology in the liver.46 In a study examining the neuroprotective effects of DMF following oxidative stress from hydrogen peroxide, DMF significantly upregulated gene expression of SOD2 in rat neural stem/progenitor cells.47 The positive results obtained with pharmacological activation of the Nrf2 antioxidant pathway, and specifically upregulation of SOD2, holds promise for the development of Nrf2 activators such as DMF for clinical use in SCD.
SOD2 and vascular function
SOD2 has been shown to be a vital enzyme in the maintenance of vascular function. Low levels of intracellular and extracellular hydrogen peroxide and superoxide contribute to physiological biochemical processes such as defense against microorganisms and cell signaling.48,49 In diseases such as SCD, hypertension, and diabetes, where there is an excess of superoxide, regulation of the SOD enzymes plays an important role in attenuating oxidative damage to the mitochondria and endothelium. Specifically, adjustments in levels of superoxide have been shown to modulate cell growth, apoptosis, inflammation, gene expression, and vascular tone via oxidation of signaling molecules and activation of redox-sensitive transcription factors.27 SOD2 overexpression and excess generation of hydrogen peroxide has been implicated in angiogenesis through increased oxidation of phosphatase and tensin homolog deleted from chromosome 10,50 cell differentiation through prolonged extracellular signal-regulated kinase 1/2 signaling,51 and pulmonary hypertension through the activation of hypoxia inducible factor 1α-O2–sensitive pathways.52 A deficiency of SOD2 in apolipoprotein E–deficient mice results in mitochondrial damage and impaired vascular relaxation in response to acetylcholine.53 The endothelial and mitochondrial damage resulting from SOD2 deficiency could not be rescued by 4,5-dihydroxy-1,3-benzene disulfonic acid–mediated scavenging of superoxide, supporting the vital role of SOD2 in maintaining vascular function.53 In vascular aging, disruption of vascular homeostasis and functional deterioration results from imbalances between pro- and antioxidants. This vascular aging-related dysfunction is more pronounced in heterozygous SOD2 mice and is associated with increased levels of mitochondrial ROS production and mitochondrial damage.54 It has been noted that SCD patients have reduced peripheral blood SOD2 expression.55 In SCD, increased oxidative scavenging of the vasodilator NO is likely a contributory factor in accelerating deterioration of vascular function.
SOD2 expression and activity must be maintained at a balance in order to avoid (1) excess superoxide and (2) overproduction of hydrogen peroxide.56 Excess superoxide disrupts cellular activity by reacting with NO and targeting iron sulfur clusters,57-59 which can impair NO-mediated vasodilation,3 activate mitochondrial uncoupling proteins, and modify complex I of the electron transport chain for diminished proton motive force and decreased adenosine triphosphate production.57,58,59 On the other hand, increased hydrogen peroxide through the upregulation of SOD2 stimulates pro-oxidants involved in apoptosis,57 targets thiol groups on proteins, and provides a substrate for oxidant production by Fenton chemistry. The deleterious consequences of abnormal SOD2 activity highlight the importance of balance in maintaining healthy vascular function and the need for careful patient selection when considering SOD2 therapies.
SOD2, reactive species, and SCD
In SCD, ischemia-reperfusion increases ROS and RNS generation through the upregulated enzymatic activity of xanthine oxidase,60-62 NADPH oxidases,63-65 and NO synthase, both coupled and uncoupled.66 Limited oxygen availability during ischemia-reperfusion impairs metabolism and increases mitochondrial ROS production.67 Electron transfer between heme iron and oxygen3 occurs at a faster rate in HbS than non-HbS,68,69 predisposing sickle RBCs to auto-oxidation and lysis. Hemolysis releases excess amounts of hemoglobin into plasma, which saturate the scavenging molecules haptoglobin and CD163.70,71 Unscavenged hemoglobin is free to react with hydrogen peroxide, forming hydroxide radicals via Fenton chemistry. It also scavenges NO, limiting its bioavailability.70,72 In the proinflammatory milieu of SCD, sickled RBCs have increased adhesion to the vascular endothelium via vascular adhesion molecule 1, intracellular adhesion molecule 1, E-selectin, and P-selectin.3,73-75 RBC adhesion to vascular endothelium activates the NF-κB pathway and increases oxidative stress as measured by TBARS formation.76 SOD and catalase were shown to reduce expression of vascular adhesion molecule 1, decreasing oxidative stress as measured by TBARS, in cultured human umbilical vein endothelial cells.76 SOD2 messenger RNA levels are reduced in the peripheral blood of SCD patients, affecting SOD2 protein levels that correlate with RBC count, reticulocyte count, platelet count, C-reactive protein, ferritin, brain natriuretic peptide values, and echocardiographic indications of pulmonary hypertension.55 Considering the beneficial effect of SOD supplementation on oxidative stress and adhesion in vitro and the deficiency of SOD2 in patients with SCD, the potential for SOD therapy in SCD takes on greater meaning. While SOD2 supplementation has the potential to offer great benefit for those SCD patients deficient in SOD2, it is important to note that SOD2 overexpression upregulates angiogenesis through hydrogen peroxide–meditated oxidation of phosphatase and tensin homolog deleted from chromosome 10.50 Angiogenic mediators such as vascular endothelial growth factor, placental growth factor, and erythropoietin are reportedly elevated in SCD.77 Given the role of SOD2 in promoting angiogenesis and the pathological angiogenesis that contributes in some cases to sickle pathology,77 it is vital to screen potential recipients of SOD2 therapy for markers of increased angiogenesis such as vascular endothelial growth factor, placental growth factor, and erythropoietin in order to avoid propagating pathological angiogenesis.
The function of SOD2 has been well studied in global and cell-type–specific knockout models. In mice, complete global deficiency of SOD2 results in early lethality with the development of severe cardiomyopathy, anemia, and neural degeneration.78 In order to study the effect of SOD2 deficiency in hematopoietic cells in vivo over long periods of time, Friedman et al transplanted murine fetal liver stem cells deficient in SOD2 into γ-irradiated metabolically normal mice.79 Sod2−/− liver stem cells were able to rescue irradiated mice, but unlike Sod2+/− and Sod2+/+ liver stem cells, erythrocyte development was impaired, and mice were persistently anemic.79 SOD2 deficiency resulted in an increased accumulation of oxidized RBC protein, decreased RBC survival, and decreased RBC membrane deformability.79 Increased rate of hemoglobin oxidation, increased heme degradation products, and reduced deformability of RBCs80 were also found in a hematopoietic stem cell transplant model of SOD2 deficiency. All of these RBC pathologies are characteristic of hemolytic anemias such as SCD,79 providing further evidence of the antioxidant’s vital role in SCD and other hemolytic anemias. Interestingly, while Friedman et al have shown that SOD2 deficiency is detrimental for RBCs, SOD2 seems nonessential for platelet functioning. Fidler et al observed increased mitochondrial ROS in a platelet-specific SOD2 knockout mouse model, but the increase in ROS did not affect platelet activation.81 Even in pathological platelet-centered immune mechanisms, such as sepsis and autoimmune inflammatory arthritis, there was no phenotypic difference between the SOD2 knockout and wild-type models.81 These studies indicate that RBCs and not platelets would be a target for therapies centered on SOD2 in SCD.
Several SOD2 gene polymorphisms and their clinical implications have been described.30,82,83 A valine-16 to alanine (Val-16-Ala) polymorphism associates with clinical complications for SCD patients. This polymorphism is correlated with decreased SOD2 activity as well as increased vaso-occlusive crises and acute splenic sequestration in children with SCD.84 Decreased SOD2 enzymatic function with the Val-16-Ala polymorphism has also been observed in cryopreserved human hepatocytes85 and isolated human erythrocytes.86 The Val-16-Ala polymorphism occurs at a high frequency among various ethnic groups: 0.4521 in Africans, 0.6492 in Latinos, 0.5375 in South Asians, 0.5162 in Europeans (non-Finnish), 0.1596 in East Asians,87 and 0.4902 in others. The high frequency of the Val-16-Ala SOD2 polymorphism and reported associations with sickle complications emphasizes the significant role that Val-16-Ala may play in contributing to the phenotypic spectrum of SCD. Recently, Ganini et al have shown that when cells and animals are exposed to a high iron/manganese ratio, SOD2 incorporates iron more so than manganese, creating a pro-oxidant peroxidase.88 This aberrant iron-bound SOD2 leads to oxidative stress and mitochondrial dysfunction both in vitro and in vivo.88 In SCD, frequent blood transfusions, decreased RBC survival, and increased intravascular hemolysis increase iron levels,89 thus increasing the iron/manganese ratio. The role of endothelial SOD2 in SCD has yet to be established. Further investigation into the Val-16-Ala SOD2 polymorphism’s antioxidant capacity and how the potential incorporation of iron into SOD2 may contribute to different sickle pathologies (eg, pulmonary hypertension, stroke, renal failure, and leg ulcers) and response to therapeutics will aid in the development of more personalized clinical care.
SOD2 and other blood disorders
While the implications of SOD2 regulation in hemolytic disorders is still a new area of investigation, there is an abundance of literature investigating how redox status, and in particular SOD2 expression and activity levels, play a role in response to pharmacological treatment of leukemias and lymphomas. In adult patients with acute lymphoblastic leukemia, the Val-16-Ala SOD2 polymorphism is associated with increased hepatotoxicity following asparaginase-based treatment.90 This increased hepatoxicity with the Val-16-Ala polymorphism suggests that asparaginase-based therapies should be used with caution in acute lymphoblastic leukemia patients with the polymorphism. In cell line models of acute myeloid leukemia (AML), treatments targeting ROS production and expression levels of SOD2 have shown efficacy.91,92 Specifically, disulfiram/copper was able to increase ROS in AML cell lines by inducing SOD2 and suppressing catalase, therefore increasing hydrogen peroxide production.91 This suggests that SOD2 induction-based therapy may be effective as an AML therapeutic. A phase 2 clinical trial investigating the use of imexon for relapsed follicular and aggressive lymphomas revealed that redox-related genes such as glutathione peroxidase and SOD2 correlate with clinical responses.93 This indicates that analysis of pretreatment markers of oxidative stress may aide in identifying patients most likely to benefit from treatment with Imexon.93 These studies show that SOD2 holds promise as a prognostic biomarker in leukemia and lymphoma and suggest it may play a role in the pathogenesis of hematologic malignancies.
Antioxidants in therapeutics
Despite the evidence of increased ROS in disease processes, the use of antioxidants in therapeutics is complicated by the necessity of maintaining physiological levels of oxygen and free radicals.94 The current status and future directions of the clinical use of antioxidant drugs have been reviewed elsewhere.95 Briefly, excitement in the 1980s and 1990s surrounding the use of antioxidant therapies to modulate disease processes was dampened as the newly developed drugs struggled to make it past clinical trials. The few notable antioxidants that have been approved for clinical use include N-acetylcysteine for acetaminophen overdose and dry eye syndrome, α-lipoic acid for diabetic nephropathy, and the flavonoids baicalein and catechins for use in osteoarthritis.95 Explanations for why antioxidant therapy has not been as resounding a success as it was hoped to be include the following: (1) oxidative stress may not be the only or the primary cause of the disease; (2) patients may not equally benefit from antioxidant therapy; (3) the target is not well selected; and (4) the antioxidant chosen may have poor selectivity for the target.95 It is then crucial for more focused research to describe how specific antioxidants play a role in modulating particular diseases.
There are currently no studies investigating the benefits of SOD2 therapy in SCD. The use of SOD2 targeted therapeutics has, however, been studied for >30 years. Therapies aimed at alleviating oxidative stress and superoxide levels through increasing SOD2 have been studied in inflammation reduction, protection from irradiation, transplantation immunosuppression, and fibrosis.96-98 In 1983, it was shown that in patients undergoing radiation, lysosome-encapsulated SOD was able to prevent inflammation and fibrosis.96 In 1994, IV administration of human recombinant SOD decreased the number of acute and chronic rejection events in recipients of cadaveric renal transplants. More recently, in 2013, oral administration of human recombinant SOD2 reduced liver fibrosis and portal pressure in cirrhotic rats.97,98 Administration of SOD2 as a drug has been complicated by the short 6-minute half-life of the enzyme due to rapid renal clearance.99 In order to circumvent this limitation, SOD2 plasmid-containing liposomes100 and small-molecule mimetics101,102 are being developed. The recent availability of improved formulations of SOD2 extends its therapeutic potential; this, along with the history of efficacy, paves the way for clinical trials in SCD.
Summary and opportunities moving forward
Increased ROS and decreased antioxidant capacity in SCD overwhelms RBCs and mitochondria and damages endothelial cells and tissues, promoting pathology. SOD2, a significant component of the enzymatic antioxidant defense system, is both transcriptionally and posttranscriptionally regulated. Upregulation of SOD2 in SCD comes with increased ROS, though this response is impaired due to decreased Nrf2 activation and posttranslational downregulation of SOD2 by RNS. The importance of increasing the antioxidant response, in particular SOD2, is underscored by increased clinical complications found with the decreased SOD2 activity of the Val-16-Ala polymorphism.84 The role of SOD2 in maintaining vascular homeostasis54 also contributes to the vital part it plays in modulating SCD pathology. Further exploration of the events that lead to SOD2 activity regulation in SCD will provide a clearer understanding of SOD2’s role in the progression of SCD pathology and potentially produce improvement in clinical outcomes. In particular, the importance of SOD2 in the vascular wall has not been thoroughly investigated. We anticipate that SOD2 supplementation would confer protection to the endothelium during acute and chronic oxidative stress events, potentially reducing the incidence of pulmonary hypertension associated with SCD.
In this era of precision medicine, it is important to consider disease in context of the patient and his or her own innate antioxidant defense systems. Increased oxidative stress is commonly found in a wide array of diseases, and the antioxidant response between patients is not uniform. Investigation of the patient’s own antioxidant expression levels and known polymorphisms that may alter activity levels will lead to more informed clinical decision making.
Acknowledgments
The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about. They also thank Heidi Schmidt for reading and editing the manuscript and Anita Impagliazzo for drawing the schematic.
Funding was provided, in whole or in part, by the National Institutes of Health, National Heart, Lung, and Blood Institute (grants R01 HL 133864 and R01 HL 128304), the American Heart Association (established investigator grant 19 EIA34770095), the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania (A.C.S.), and the American Society of Hematology Minority Medical Student Award Program (A.M.D.-O.).
Authorship
Contribution: A.M.D.-O. and A.C.S. conceived the idea; A.M.D.-O. wrote the manuscript; K.C.W., E.M.N., and A.C.S provided direction and extensive edits; and all authors approved the final version for submission.
Conflict-of-interest disclosure: A.C.S. receives research funding from Bayer Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Adam C. Straub, Department of Pharmacology and Chemical Biology, The Heart, Lung, Blood and Vascular Medicine Institute, University of Pittsburgh School of Medicine, E1254 Biomedical Science Tower, 200 Lothrop St, Pittsburgh, PA 15216; e-mail: astraub@pitt.edu.

