

Key Points
We report longitudinal mutational analyses of 2 patients with high-risk MDS and AML experiencing spontaneous disease remissions.
Both patients had persistent clonal hematopoiesis during remission, harboring all but 1 of the mutations from the initial diagnostic sample.
Introduction
Spontaneous remission (SR) of myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) in the absence of disease-modifying therapy is rare.1,,,-5 One review identified 46 cases of AML SRs published between 1950 and 2014.6 Infections preceded >70% of SRs, leading to the hypothesis that the immunostimulatory effect of severe infections may occasionally induce immune-mediated blast clearance. Others suggested that donor lymphocytes from nonirradiated blood products may induce SR through allo-immune effects.7,-9 However, infections and transfusion-dependent cytopenias are common in AML patients, and thus their association with SRs may be coincidental.4 SR has also been documented in MDS,10 particularly in children and young adults with abnormalities of chromosome 7.11,,,-15
Over the past 10 years, next-generation sequencing (NGS) techniques led to the identification of novel recurrently mutated genes in myeloid neoplasia (MN), expanding our knowledge about the genetic basis of MN and evolution of premalignant clonal hematopoiesis to MDS or AML. Detailed mutational analyses of patients achieving SR of MN have not yet been reported. Here, we describe the clinical course and sequential genetic analyses of 2 patients with high-risk MDS and AML who achieved SRs.
Case description
Patient 1
A 59-year-old man with no known history of hematologic disease was evaluated for a suspected rheumatologic disorder. He reported pain in his small joints, night sweats, and generalized exanthema that had started 3 weeks earlier and had improved with low-dose oral steroids and brine bath therapy. Laboratory tests showed neutropenia, mild anemia, and thrombocytopenia (Figure 1; supplemental Table 1). A BM aspirate was hypocellular with dysplastic erythropoiesis and 3% myeloblasts, consistent with a diagnosis of MDS, with a normal karyotype. On histologic examination of a BM trephine biopsy, 30% to 35% CD34+/CD117+ blasts were noted. A repeat BM aspiration (day 18) revealed 18% myeloblasts, and a diagnosis of MDS with excess blasts was made. The patient had stable blood counts, and a search for a donor for an allogeneic stem cell transplantation (ASCT) was initiated. At follow-up visits 5 and 8 weeks later, his blood counts had normalized, and he continued to receive low-dose oral corticosteroids for polyarthritis with proximal interphalangeal joint and ankle pain. At the second visit (day 75), BM aspiration and biopsy showed normal trilineage hematopoiesis without dysplasia or excess blasts. Cytogenetic analysis was not repeated. Seven months after initial presentation, leukopenia and anemia recurred, and a BM aspirate showed 58% myeloblasts. Cytogenetic analysis revealed a tetraploid clone with a 5q deletion. The patient received sequential high-dose cytarabine and mitoxantrone induction chemotherapy,16 achieved complete remission (CR), and subsequently underwent ASCT from an HLA-matched donor. He is in ongoing CR 21 months after transplantation.

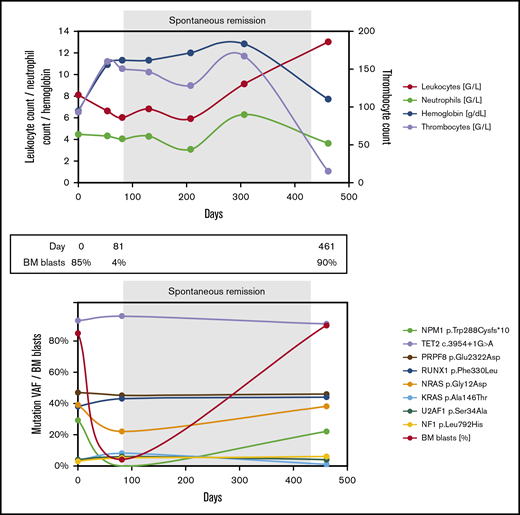
Blood counts and genetic landscape during the disease course of patient 1. BM, bone marrow; NA, not available; SCT, stem cell transplantation; VAF, variant allele frequency.
Blood counts and genetic landscape during the disease course of patient 1. BM, bone marrow; NA, not available; SCT, stem cell transplantation; VAF, variant allele frequency.
Patient 2
A 72-year-old man was hospitalized for decompensated congestive heart failure. Laboratory testing revealed anemia and thrombocytopenia (Figure 2A; supplemental Table 2). BM aspiration showed marrow infiltration by 85% myeloblasts, and the patient was diagnosed with AML with a normal karyotype and mutated NPM1. Three skin lesions on the abdomen and back were suggestive of extramedullary disease, but skin biopsy was not performed. The patient suffered from multiple comorbidities, including ischemic and valvular heart disease and chronic kidney disease. He decided against initiation of antileukemic therapy and opted for best supportive care. Over the next few weeks, he received 5 units of packed red blood cells. Seven weeks after initial presentation, histologic examination of a skin lesion showed infiltration by myeloid blasts. Soon thereafter, his blood counts improved spontaneously, he became transfusion independent, and his skin lesions disappeared without antileukemic treatment. Eleven weeks after the initial presentation, repeat BM aspiration and biopsy showed no evidence of AML, and quantitative polymerase chain reaction (qPCR) testing for mutated NPM1 was negative. More than 1 year later, the patient was readmitted with weakness and bicytopenia. BM examination confirmed AML recurrence, which tested positive for the NPM1 mutation and now displayed an abnormal karyotype. The patient reaffirmed his decision to forego antileukemic treatment and died a few days later.
Methods
Results and discussion
At initial presentation, patient 1 carried 2 distinct RUNX1 mutations and mutations in SRSF2, IDH2, and BCOR (Figure 1; supplemental Table 1). During his SR, BM (day 75) and peripheral blood (day 168) samples tested negative for 1 of the RUNX1 mutations found at initial presentation, but the second RUNX1 mutation and mutations in SRSF2, IDH2, and BCOR were still detected in the BM, albeit at lower allele frequencies. At the time of disease recurrence (day 220), the spectrum of gene mutations in BM was identical to that in the primary presentation, with reappearance of the second RUNX1 mutation. Cytogenetic analysis showed an evolved karyotype with gain of a 5q deletion. After induction chemotherapy (day 299), the patient again reached CR. At that time, 1 RUNX1 mutation and the SRSF2 mutation persisted, whereas the other mutations became undetectable. On day 360 and day 577 (30 and 247 days after ASCT), no mutation was detected consistent with full donor chimerism and ongoing CR.
In patient 2, 8 different gene mutations were identified at the time of primary diagnosis (Figure 2A; supplemental Table 2). During his SR (day 81), the NPM1 mutation became undetectable in BM by NGS and by qPCR (sensitivity, <10−5). All other mutations persisted despite the normalized blood counts and BM morphology. At disease recurrence (day 461), all mutations found initially were detected, including the NPM1 mutation, and there was clonal evolution on the cytogenetic level.
Neither of our 2 patients presented with fever or severe infection, which have previously been implicated as triggers of immune-mediated SR in AML. Patient 1 did not require transfusions before achieving SR, and patient 2 received only irradiated blood products, which made an allo-immune mechanism unlikely. SR has also been reported in AML patients receiving high-dose corticosteroids.20 Patient 1 started taking low-dose prednisolone before his initial diagnosis of MDS/AML, but we cannot exclude that direct or immunomodulatory effects of steroids contributed to the SR.21
We found that both our patients, on the genetic level, did not reconstitute normal polyclonal hematopoiesis during their clinically defined SR. Instead, both had persistent clonal hematopoiesis that shared most somatic mutations with the malignant clone. In fact, in each patient, only 1 driver mutation found in the AML/MDS specimen became undetectable at the time of SR, while all other mutations persisted. This finding illustrates that premalignant hematopoietic clones can sequentially acquire multiple mutations, yet still sustain normal-appearing hematopoiesis. Gain of a single mutation might then be sufficient to tip the balance and transform clinically asymptomatic clonal hematopoiesis into full-blown MN. The close genetic relationship between the AML/MDS clone and its immediate premalignant ancestor might also explain why, under certain rare circumstances, the premalignant clone can regain a selective advantage over the AML/MDS clone. Although the mechanisms that allowed re-expansion of preleukemic clones in our patients are uncertain, previous case reports suggest that altered cytokine levels or immune function might play a role.
In summary, we report, for the first time, longitudinal mutational profiling of patients with AML or high-risk MDS who achieved SR. Our 2 patients had persistent clonal hematopoiesis during SRs, in both cases lacking 1 single mutation compared with the malignant clone. This report illustrates the dynamic, and sometimes nonlinear, clonal evolution of MN and provides insights into the genetic mechanisms underlying SR.
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by a clinical research fellowship from the European Hematology Association, and by a grant (DRG SFB 1243, TP A06) from the German Research Council.
Authorship
Contribution: V.V.G., K.S., M.R.-T., and K.H.M coordinated the analyses; M.H., X.S., M.S., M.F., W.H., and K.S. were involved in clinical patient care; V.V.G., M.R.-T. and K.H.M. generated and analyzed sequencing data; A.D., S.S., M.N., M.S., M.F., K.S., and K.H.M. performed other diagnostic testing; V.V.G, M.R.-T., and K.H.M drafted the manuscript; and all coauthors read and approved the final version of the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Klaus H. Metzeler, Laboratory for Leukemia Diagnostics, Department of Internal Medicine III, University Hospital, LMU Munich, Marchioninistr 15, 81377 Munich, Germany; e-mail: klaus.metzeler@med.lmu.de.

