

Key points
T cells from patients early in myeloma therapy exhibit better fitness for CAR T manufacturing than those from relapsed/refractory patients.
CAR T cells may be more effective if manufactured from patients before onset of relapsed/refractory disease.
Introduction
Chimeric antigen receptor (CAR) T cells are a promising, emerging therapy for multiple myeloma. CAR T cells directed against the B-cell maturation antigen (BCMA) have demonstrated impressive initial results, but available data suggest that most patients with initial responses eventually progress.1,,-4 New strategies are therefore needed to improve CAR T-cell therapy for multiple myeloma.
Autologous CAR T-cell efficacy depends on the functional capacity of patients’ endogenous T cells. We recently reported an analysis of patients with chronic lymphocytic leukemia treated with anti-CD19 CAR T cells to identify predictors of clinical response. Among all baseline disease- and patient-specific parameters analyzed, frequency of a memory T-cell subset, defined by a CD8+ CD45RO− CD27+ immunophenotype, in the premanufacturing leukapheresis product was the only parameter identified to be significantly associated with clinical response.5 Frequency of this memory subset in the leukapheresis product was associated with transcriptomic and metabolomic features of early memory differentiation and enhanced antigen-responsive cytotoxicity of the manufactured product. Similarly, in our phase 1 trial of anti-BCMA CAR T cells (CART-BCMA) for multiple myeloma, higher frequency of CD8+ CD45RO− CD27+ T cells and higher CD4/CD8 ratio at time of leukapheresis were the only factors associated with clinical response among all patient- and disease-specific parameters analyzed.3 Understanding how the CD8+ CD45RO− CD27+ T-cell phenotype and CD4/CD8 ratio vary among patients with multiple myeloma could help identify the optimal clinical setting for T-cell collection and subsequent CAR T-cell manufacturing.
Multiple myeloma is associated with deficiencies in T-cell immunity,6,7 and many multiple myeloma therapies are toxic to lymphocytes. We therefore hypothesized that the frequency of T cells with the CD8+ CD45RO− CD27+ phenotype and the CD4/CD8 ratio would be higher in multiple myeloma patients early in the disease course, when disease burden is low and prior exposure to therapy is minimal, compared with the relapsed/refractory disease setting. We evaluated this hypothesis in a unique set of leukapheresis samples from patients with multiple myeloma who underwent leukapheresis prior to first-line autologous stem cell transplant (ASCT), after response to induction therapy (postinduction), and expanded with anti-CD3/anti-CD28 agonistic monoclonal antibody–conjugated beads at clinical scale, mirroring the procedure used in many CAR T-cell manufacturing processes. We compared the leukapheresis product features and magnitude of ex vivo expansion from this postinduction sample set with those of patients with relapsed/refractory multiple myeloma who participated in our phase 1 trial of anti-CART-BCMA and underwent leukapheresis for CART-BCMA manufacturing on this trial.
Methods
The postinduction cohort consisted of 38 subjects who participated in previously reported8,,,-12 clinical trials (clinicaltrials.gov identifiers #NCT01245673, #NCT01426828, and #NCT00499577). On these prior trials, leukapheresis was performed after response to initial multiple myeloma therapy and vaccine priming and just before consolidation with high-dose chemotherapy and ASCT; cells then underwent ex vivo expansion with anti-CD3/anti-CD28 beads and were reinfused after ASCT to assess effects of autologous T-cell infusion on post-ASCT immune reconstitution. The relapsed/refractory cohort consisted of 25 patients who received CART-BCMA cells, which were manufactured using a similar anti-CD3/anti-CD28 monoclonal antibody bead expansion protocol as the postinduction cohort, on a recently reported phase 1 clinical trial3 ; in this study, leukapheresis was performed just after enrollment following a 2-week washout from prior myeloma therapy. Cryopreserved leukapheresis samples were analyzed by flow cytometry for CD4/CD8 ratio and the proportion of T cells exhibiting the CD8+ CD45RO− CD27+ memory immunophenotype as previously described.5 Growth curves from the clinical T-cell cultures were used to calculate the number of population doublings by day 9 (PD9) as a measure of proliferative capacity. Insufficient data were available to calculate PD9 in 6 subjects from the postinduction cohort and 4 subjects from the relapsed/refractory cohort. Associations between cohort and continuous variables were assessed using the Wilcoxon rank-sum test. Associations between continuous variables were evaluated using Spearman correlations. Clinical specimens and data were collected on institutional review board–approved protocols.
Results and discussion
Table 1 depicts clinical features of the postinduction and relapsed/refractory cohorts. The cohorts were similar in age. The postinduction cohort had shorter time since multiple myeloma diagnosis to enrollment (median 222 days vs 4.6 years), fewer prior lines of therapy (median 1 vs 7), and less bone marrow cellularity occupied by myeloma plasma cells (median 13% vs 65%) at time of leukapheresis.
Cohort characteristics
| Postinduction (N = 38) | Relapsed/refractory (N = 25) | |
|---|---|---|
| Age, median (range), y | 55 (41-68) | 58 (44-75) |
| Time since MM diagnosis, median (range) | 222 d (76-783) | 4.6 y (1.8-14.5) |
| High-risk cytogenetic features, % | 40 (only 20 with available data) | 96 |
| Lines of prior MM therapy, median (range) | 1 (1-4) | 7 (3-13) |
| Prior treatment exposure, median (range), % | ||
| Thalidomide, lenalidomide, or pomalidomide | 76 | 100 |
| Proteasome inhibitor | 55 | 100 |
| Alkylating agent | 21 | 100 |
| High-dose chemotherapy + ASCT | N/A* | 92 |
| Bone marrow cellularity occupied by MM at time of enrollment, median (range), % | 13 (0-80) | 65 (0-95) |
| Postinduction (N = 38) | Relapsed/refractory (N = 25) | |
|---|---|---|
| Age, median (range), y | 55 (41-68) | 58 (44-75) |
| Time since MM diagnosis, median (range) | 222 d (76-783) | 4.6 y (1.8-14.5) |
| High-risk cytogenetic features, % | 40 (only 20 with available data) | 96 |
| Lines of prior MM therapy, median (range) | 1 (1-4) | 7 (3-13) |
| Prior treatment exposure, median (range), % | ||
| Thalidomide, lenalidomide, or pomalidomide | 76 | 100 |
| Proteasome inhibitor | 55 | 100 |
| Alkylating agent | 21 | 100 |
| High-dose chemotherapy + ASCT | N/A* | 92 |
| Bone marrow cellularity occupied by MM at time of enrollment, median (range), % | 13 (0-80) | 65 (0-95) |
MM, multiple myeloma; N/A, not applicable.
Patients in the postinduction cohort underwent leukapheresis prior to ASCT.
The postinduction cohort exhibited a significantly higher percentage of T cells with the CD8+ CD45RO− CD27+ memory phenotype (median 43.9% vs 29.0%, P = .001; Figure 1A) and significantly higher CD4/CD8 ratio (median 2.6 vs 0.87, P < .0001; Figure 1B) compared with the relapsed/refractory cohort. We also compared the postinduction cohort to the subset of the relapsed/refractory cohort that exhibited at least partial response to CART-BCMA (N = 12), as we previously reported that the CD8+ CD45RO− CD27+ percentage and CD4/CD8 ratio were higher among CART-BCMA responders.3 The median percentage of T cells with the CD8+ CD45RO− CD27+ memory phenotype was higher in the postinduction cohort compared with CART-BCMA responders, but the difference was of only borderline statistical significance (median 43.9% vs 33.1%, P = .07; Figure 1A). The median CD4/CD8 ratio was significantly higher in the postinduction cohort compared with CART-BCMA responders (median 2.6 vs 1.3, P = .0009; Figure 1B). T cells from the postinduction cohort exhibited significantly higher capacity for ex vivo proliferation during manufacturing, as indicated by PD9, compared with the overall relapsed/refractory cohort (median 5.3 vs 4.5, P = .0008) and the CART-BCMA responders (median 5.3 vs 4.6, P = .009; Figure 1C).
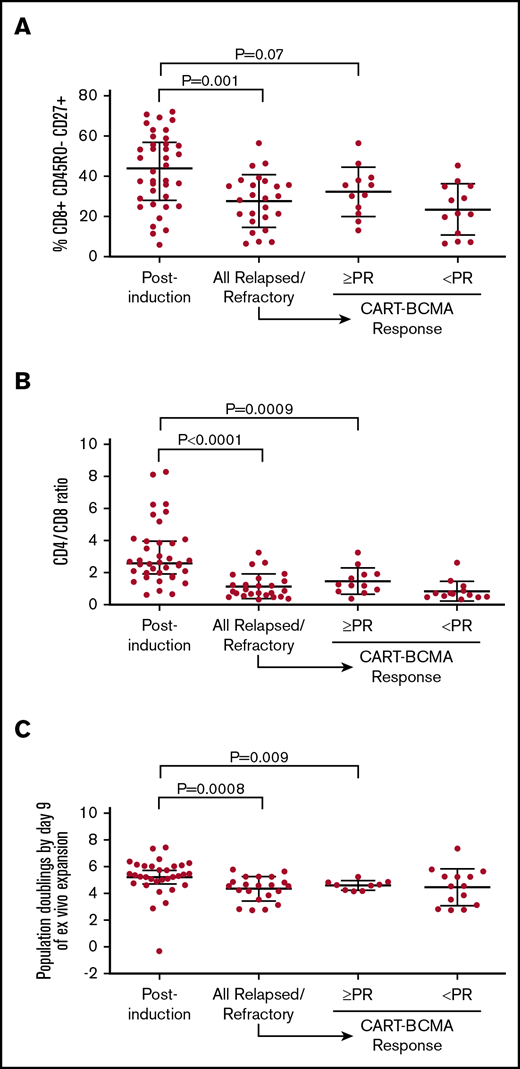
Comparison of apheresis samples in postinduction and relapsed/refractory cohorts. Percent of T cells with the CD8+ CD45RO− CD27+ memory phenotype (A), CD4/CD8 ratio (B), and PD9 of ex vivo stimulation (C) with agonistic anti-CD3/anti-CD28–conjugated microbeads. Rightmost 2 columns in each graph depict each parameter in the relapsed/refractory cohort, separated according to response to CART-BCMA. In each analysis, the postinduction cohort was compared with the overall relapsed/refractory cohort and to the subset of the relapsed/refractory cohort that achieved at least partial response (PR) to CART-BCMA.
Comparison of apheresis samples in postinduction and relapsed/refractory cohorts. Percent of T cells with the CD8+ CD45RO− CD27+ memory phenotype (A), CD4/CD8 ratio (B), and PD9 of ex vivo stimulation (C) with agonistic anti-CD3/anti-CD28–conjugated microbeads. Rightmost 2 columns in each graph depict each parameter in the relapsed/refractory cohort, separated according to response to CART-BCMA. In each analysis, the postinduction cohort was compared with the overall relapsed/refractory cohort and to the subset of the relapsed/refractory cohort that achieved at least partial response (PR) to CART-BCMA.
Both the proportion of CD8+ CD45RO− CD27+ T cells and the CD4/CD8 ratio varied considerably within the postinduction cohort. Within this cohort, we did not identify any association between these parameters and prior exposure to particular therapeutic classes, elapsed time between multiple myeloma diagnosis and leukapheresis, or degree of bone marrow plasma cell infiltration at time of leukapheresis (as a measure of myeloma burden; supplemental Table). A larger sample or molecular characterization of multiple myeloma cells might identify myeloma- or treatment-related factors that account for the heterogeneity in T-cell parameters observed in the postinduction cohort.
Our results suggest that CAR T cells manufactured from leukapheresis samples obtained after response to induction therapy would be, on average, more clinically effective than those obtained from heavily relapsed/refractory multiple myeloma patients. One strength of our study is the unique sample set from relapsed/refractory patients treated with CART-BCMA and postinduction patients who underwent a clinical-scale ex vivo T-cell expansion, allowing comparison of not only leukapheresis phenotype but also clinical-scale expansion potential. Examination of T-cell phenotypes at additional intermediate time points between postinduction and heavily relapsed/refractory settings might refine the optimal window for T-cell collection. Although unlikely, comparisons between the groups could also have been confounded by vaccines administered in the postinduction but not in the relapsed/refractory cohort. In these regards, our findings are hypothesis generating and provide rationale to evaluate the potency of CAR T cells generated from patients with multiple myeloma at different points in the disease course and from the CD45RO− CD27+ memory subset. Now that safety of anti-BCMA CAR T cellshas been demonstrated, clinical evaluation of CAR T cells in earlier settings would be justified for high-risk patients, who typically respond well to first-line therapy but progress quickly and have a poor prognosis even with modern therapy. Alternatively, measures could be taken in the relapsed/refractory setting to modify the leukapheresis product to overcome its deficiencies. These approaches are being evaluated in ongoing clinical trials.
Original data can be obtained by contacting the corresponding author.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the staff of the Penn Cell and Vaccine Production Facility and the nurses and physicians in the apheresis unit of the Hospital of the University of Pennsylvania.
This work was supported by National Institutes of Health, National Cancer Institute award P01 CA214278. A.L.G. is a Scholar in Clinical Research of the Leukemia and Lymphoma Society and additionally acknowledges support from a Conquer Cancer Foundation Career Development Award.
Authorship
Contributions: A.L.G. designed research, performed research, collected data, analyzed/interpreted data, and wrote the manuscript; E.K.D., A.D.C., M.M.D., B.L.L., D.L.S., E.A.S., D.T.V., A.W., A.P.R., M.C.M., C.H.J., and J.J.M. designed research, performed research, collected data, and analyzed/interpreted data; W.-T.H. analyzed/interpreted data; and J.A.F. designed research and analyzed/interpreted data.
Conflict-of-interest disclosure: A.L.G. receives research funding from Novartis, Amgen, Janssen, and Tmunity; receives consultancy fees from Kite Pharma and Surface Oncology; and has intellectual property licensed by the University of Pennsylvania to Novartis. A.D.C. receives research support and consulting fees from Bristol-Meyers Squibb and consulting fees from Celgene, Kite Pharma, Janssen, Seattle Genetics, Oncopeptides, Takeda, Array Biopharma, GlaxoSmithKline; and has intellectual property licensed by the University of Pennsylvania to Novartis. J.A.F. has patents related to CAR T-cell therapy and receives research funding from Novartis Pharmaceuticals Corporation and Tmunity Therapeutics. M.M.D. has intellectual property licensed by the University of Pennsylvania to Novartis. B.L.L. serves on Scientific Advisory Boards for Avectas, Brammer Bio, Cure Genetics, Incysus, and Vycellix, receives consulting fees from CRC Oncology, has licensed intellectual property and royalties from Novartis, and is cofounder, equity holder, and has licensed intellectual property from Tmunity Therapeutics. D.L.S. receives research funding from Tmunity and serves on the Scientific Advisory Board for Poseida Therapeutics. E.A.S. receives research funding from AbbVie and personal fees from Celgene, Takeda, Janssen, and Amgen and has intellectual property licensed by the University of Pennsylvania to Novartis. D.T.V. receives personal fees from Karyopharm, Amgen, Millennium/Takeda, Celgene and research funding from GSK. M.C.M. receives research funding from Novartis and has intellectual property licensed by the University of Pennsylvania to Novartis. C.H.J. receives research funding from Novartis, is a scientific founder of Tmunity Therapeutics (founder’s stock but no income from Tmunity), and has intellectual property licensed by the University of Pennsylvania to Novartis. J.J.M. receives research funding from Novartis, Incyte, The Parker Institute and has intellectual property licensed by the University of Pennsylvania to Novartis. The remaining authors declare no competing financial interests.
Correspondence: Alfred L. Garfall, Perelman Center for Advanced Medicine, 12th Floor, South Pavilion Extension, 3400 Civic Center Blvd, Philadelphia, PA 19104; e-mail: alfred.garfall@pennmedicine.upenn.edu.

