

Key Points
Transcriptional profile of LCH CD1a+CD207+ DCs is most closely related to that of CD1c+ mDCs in the blood.
Lineage tracing with BRAFV600E and HLA-DQB2 expression supports CD1c+ mDCs as precursors to LCH CD1a+CD207+ DCs.

Abstract
Langerhans cell histiocytosis (LCH) is a myeloproliferative disorder that is characterized by the inflammatory lesions with pathogenic CD1a+CD207+ dendritic cells (DCs). BRAFV600E and other somatic activating MAPK gene mutations have been identified in differentiating bone marrow and blood myeloid cells, but the origin of the LCH lesion CD1a+CD207+ DCs and mechanisms of lesion formation remain incompletely defined. To identify candidate LCH CD1a+CD207+ DC precursor populations, gene-expression profiles of LCH lesion CD1a+CD207+ DCs were first compared with established gene signatures from human myeloid cell subpopulations. Interestingly, the CD1c+ myeloid DC (mDC) gene signature was most enriched in the LCH CD1a+CD207+ DC transcriptome. Additionally, the BRAFV600E allele was not only localized to CD1a+CD207− DCs and CD1a+CD207+ DCs, but it was also identified in CD1c+ mDCs in LCH lesions. Transcriptomes of CD1a+CD207− DCs were nearly indistinguishable from CD1a+CD207+ DCs (both CD1a+CD207low and CD1a+CD207high subpopulations). Transcription profiles of LCH lesion CD1a+CD207+ DCs and peripheral blood CD1c+ mDCs from healthy donors were compared to identify potential LCH DC-specific biomarkers: HLA-DQB2 expression was significantly increased in LCH lesion CD1a+CD207+ DCs compared with circulating CD1c+ mDCs from healthy donors. HLA-DQB2 antigen was identified on LCH lesion CD1a+CD207− DCs and CD1a+CD207+ DCs as well as on CD1c+(CD1a+CD207−) mDCs, but it was not identified in any other lesion myeloid subpopulations. HLA-DQB2 expression was specific to peripheral blood of patients with BRAFV600E+ peripheral blood mononuclear cells, and HLA-DQB2+CD1c+ blood cells were highly enriched for the BRAFV600E in these patients. These data support a model in which blood CD1c+HLA-DQB2+ mDCs with activated ERK migrate to lesion sites where they differentiate into pathogenic CD1a+CD207+ DCs.
Introduction
Langerhans cell (LC) histiocytosis (LCH) is a disorder characterized by the accumulation of pathogenic dendritic cells (DCs; LCH DCs) in inflammatory lesions. Clinical presentations are highly variable, ranging from single lesions to potentially lethal disseminated disease.1 Clinically, LCH is defined by high-risk (lesions involving liver, spleen, and/or bone marrow involvement) and low-risk disease (all other sites) based on risk of death.2 LCH lesion biopsies are characterized by CD1a+CD207+ “histiocytes” among extensive inflammatory infiltrate with common histology regardless of extent of disease or clinical risk.3,4 Mutually exclusive activating somatic mutations in MAPK pathway genes localizing to LCH lesion CD1a+CD207+ DCs have been identified in ∼85% cases, with BRAFV600E mutation as the most common.3,5-10
Despite recent advances in identifying mutations in LCH, little is known about the differentiation pathway(s) and cell(s) of origin of LCH CD1a+CD207+ DCs. Historically, pathologic LCH lesion CD1a+CD207+ DCs were proposed to arise from CD207+ epidermal LCs or along a parallel pathway.11,12 However, transcriptome analysis of LCH lesion CD1a+CD207+ DCs identified increased expression of genes associated with early myeloid differentiation compared with epidermal LCs.13-15 Furthermore, despite early reports of langerin/CD207 expression being unique to epidermal LCs, multiple alternative CD207-expressing DC populations have been discovered,16 highlighting the possibility of multiple potential differentiation pathways for the LCH lesion CD1a+CD207+ DCs. Lineage-mapping experiments identified the BRAFV600E allele in CD34+ hematopoietic stem cells and peripheral blood of patients with high-risk LCH. The BRAFV600E allele in peripheral blood mononuclear cells (PBMCs) localized primarily to myeloid populations (eg, CD11c+ cells and CD14+ cells), though in some cases, it was also detected in other lineages, including lymphoid cells.3,17 In a mouse model, enforced expression of BRAFV600E mutation in CD11c+ cells recapitulated a phenotype with features of high-risk LCH, including development of lesions with CD1a+CD207+ DCs.3 These findings suggest that pathogenic LCH lesion CD1a+CD207+ DCs may arise from myeloid cell precursors with an activating MAPK gene mutation.1,3
The pathway from CD34+ hematopoietic stem cell or early myeloid precursor to LCH lesion CD1a+CD207+ DCs is not known. Monocytes are the most abundant myeloid mononuclear cells, comprising ∼10% of PBMCs. Blood monocyte populations include CD14+CD16− “classical” monocytes and less abundant CD16+ monocytes.18,19 Classical monocytes have the ability to differentiate into tissue DCs and macrophages in vitro (reviewed in Collin and Bigley19 ). CD16+ monocytes have been further classified into 2 subpopulations based on CD16 and CD14 expression, with CD16++CD14+ “nonclassical monocytes” more abundant than CD16+CD14+ “intermediate” monocytes.18,20 Intermediate monocytes, which constitute a very small percentage in blood, appear to have both phagocytic and inflammatory functions, whereas nonclassical monocytes are more limited to inflammation with ability to produce tumor necrosis factor α (TNF-α) and interleukin 1β (IL-1β) upon activation.21 Peripheral blood DCs are rarer than monocytes (1% to 2% of PBMCs), including plasmacytoid DCs (pDCs) and myeloid DCs (mDCs).19,22,23 mDCs express antigens including CD11c, CD13, CD33, and CD11b, but lack monocyte antigens CD14 and CD16. This population may be further split into CD141+ mDCs (cDC1s) and CD1c+ mDCs (cDC2s). CD1c+ mDCs (cDC2s) are the major DCs in peripheral blood, tissues, and lymphoid cells (reviewed in Collin and Bigley,19 Collin and Bigley,22 and Collin et al23 ). CD1c+ mDCs from peripheral blood have less “activated” phenotype compared with the tissues CD1c+ mDCs (reviewed in Collin and Bigley,19 Collin and Bigley,22 and Collin et al23 ). In general, CD1c+ mDCs have low intrinsic capacity to cross-present antigen to CD8+ T cells, but they have enhanced capacity to stimulate naive CD4+ T cells.24-28 As for CD141+ mDCs, they have high capacity to cross-present antigens to CD8+ T cells, mediate efficient recognition of viral antigens, and engage necrotic cells via CLEC9A.24-27,29,30
Both CD14+ monocytes and CD1c+ mDCs may acquire a CD1a+CD207+ DC phenotype in vitro.14,15,17,31 This is in line with ontogeny of the epidermal LCs where epidermal LC precursors are formed during embryonic and fetal hematopoiesis and self-renew in the steady state, but may be replaced by phenotypically and functionally indistinguishable bone marrow–derived myeloid cells.32-35 Similarly, dermal DCs that express CD207 may arise from common DC precursors (CDPs), macrophage DC precursors (MPPs), as well as circulating hematopoietic stem cells (HSCs) and monocytes.16,36-38 Given the plasticity of mononuclear cells and the broad range of tissue distribution, there are many candidates for cell of origin and differentiation pathways leading to LCH CD1a+CD207+ DCs and associated lesion formation (Figure 1).
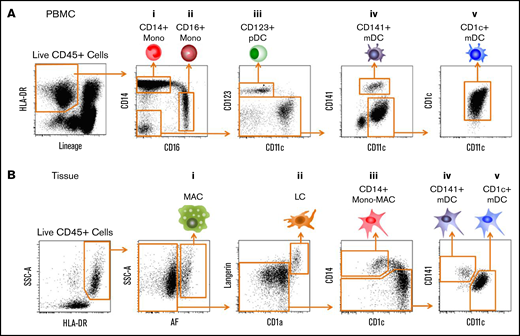
Candidate LCH precursors: myeloid lineages in peripheral blood and tissue. (A) CD45+Lineage−HLA-DR+ subsets from PBMCs (lineage contains CD3, CD19, CD20, and CD56). (i) CD14+CD16− classical monocytes. (ii) CD14lowCD16+ nonclassical monocytes. (iii) CD11c−CD123+ pDCs. (iv) CD11clowCD141+ mDCs (DC1). (v) CD11c+CD1c+ mDCs (DC2). (B) CD45+HLA-DR+ subsets from tissues. (i) AF+ macrophages. (ii) CD1a+Langerin+ LCs. (iii) CD11c+CD14+ monocytes-macrophages (Mono-Mac). (iv) CD11clowCD141+ mDCs. (v) CD11c+CD1c+ mDCs. SSC-A, side scatter area.
Candidate LCH precursors: myeloid lineages in peripheral blood and tissue. (A) CD45+Lineage−HLA-DR+ subsets from PBMCs (lineage contains CD3, CD19, CD20, and CD56). (i) CD14+CD16− classical monocytes. (ii) CD14lowCD16+ nonclassical monocytes. (iii) CD11c−CD123+ pDCs. (iv) CD11clowCD141+ mDCs (DC1). (v) CD11c+CD1c+ mDCs (DC2). (B) CD45+HLA-DR+ subsets from tissues. (i) AF+ macrophages. (ii) CD1a+Langerin+ LCs. (iii) CD11c+CD14+ monocytes-macrophages (Mono-Mac). (iv) CD11clowCD141+ mDCs. (v) CD11c+CD1c+ mDCs. SSC-A, side scatter area.
In this study, we sought to define potential origins of LCH lesion CD1a+CD207+ DCs as an opportunity to identify potential therapeutic targets for LCH and also to define the impact of MAPK pathway activation in myeloid differentiation.
Materials and methods
Subjects
This study was performed according to protocols approved by the institutional review boards of Baylor University College of Medicine, Singapore Singhealth, and National Health Care Group research ethics committees. Peripheral blood and biopsy specimens were obtained from LCH patients (supplemental Table 1a-b). LCH biopsy specimens were identified through clinical diagnosis, including CD207+ immunohistochemistry. Control skin specimens were obtained from discarded tissues from elective procedures, and control peripheral blood specimens were collected from healthy pediatric donors without blood or cancer disorders or inflammatory conditions.
Flow cytometry and cell sorting
Peripheral blood specimens from healthy donors and LCH patients as well as LCH biopsy samples were collected and processed as described previously.13 Antibodies used for LCH biopsy samples and peripheral blood specimens are listed in supplemental Table 2. Fluorescence-activated cell sorting (FACS) was performed using BD FACS Aria Fusion and BD FACSCanto and data analyzed with FCS Express 6 (De Novo Software, Glendale, CA) and FlowJo (TreeStar, Ashland, OR).
Genomic DNA analysis
For genomic DNA extraction, FACS-sorted populations were collected in ice-cold phosphate-buffered saline supplemented with 1 mM EDTA and 1% bovine serum albumin. Genomic DNA was isolated from sorted subpopulations from peripheral blood and LCH lesion specimens using QIAamp DNA micro with RNAse A treatment (Qiagen, Valencia, CA) and then amplified with the REPLI-g Midi kit per the manufacturer’s instructions. The BRAF-V600E mutation assay (Somatic Mutation Assay for BRAF_476; Qiagen) was performed as described previously3 with 30 ng of amplified genomic DNA. Triplicate reactions were performed for each sample. All experiments were performed on a CFX96 real-time system (Bio-Rad Laboratories, Hercules, CA). All samples that were used in this analysis are listed in supplemental Table 3b.
Illumina HumanHT-12 V4.0 expression Beadchip processing and analysis
Total RNA was purified from sorted subpopulations from peripheral blood and lesion specimens according to the Qiagen RNeasy Micro kit (Qiagen). RNA integrity was determined using Agilent Bioanalyzer, and the RNA integrity numbers were calculated. Biotinylated complementary RNA was prepared according to the protocol by Epicentre TargetAmp 2-Round Biotin-aRNA Amplification kit 3.0 using 500 pg of total RNA. Hybridization of complementary DNA (cDNA) was performed on Illumina Human-HT12 version 4 chips (Illumina, San Diego, CA).
Array data were extracted at the probe set level with no background subtraction using Illumina’s BeadStudio software. These raw data were then normalized by the quantile method using the lumi package in R/Bioconductor v2.13.1. A part of this data was previously reported in Haniffa et al24 and McGovern et al39 and the data set can be found in the Gene Expression Omnibus data repository (GSE35457 and GSE85305).
For generation of human myeloid subpopulation gene signatures for connectivity map (CMAP) analysis40 as previously described in Haniffa et al,24 1 cell subset was compared with other cell subsets pooled using the Student t test in R statistical software. Differentially expressed genes (DEGs) were selected with a Benjamini-Hochberg (BH) multiple testing40 corrected P < .05. CMAP analysis40 was performed comparing myeloid cell signature gene subsets with the LCH lesion CD1a+CD207+ DC gene-expression data after removal of the tissue-specific probes. The samples used in this analysis are listed in supplemental Table 3a.
Hierarchal clustering was performed by comparing the expression profiles across the set of samples using 1 − (centered) correlation for the distance metric with average linkage clustering. All samples used in this analysis are listed in supplemental Table 3a.
BubbleGUM software as described in Spinelli et al41 was used to perform multiple gene set enrichment analysis (GSEA) on all possible pairwise comparisons. A GCT file containing the preprocessed and normalized expression data were input into the BubbleGum module alongside a CLS class file, defining cell-type–specific phenotype labels associating each sample in the expression data. A GMT file containing the predefined gene signatures for CD1c+ mDCs, CD141+ mDCs, LCs, CD14+ monocyte-derived macrophages (also referred as CD14+ DCs), macrophages, CD14+ monocytes, and CD16+ monocytes, to be tested for enrichment and a CHIP file, corresponding to the CHIP platform were also included. The gene signature for each myeloid subpopulation is listed in supplemental Table 4. A weighted enrichment statistic (described in Subramanian et al42 ) was used to calculate the degree of the enrichment of each gene signature. The data were displayed as an array of circles, or a BubbleMap in which the color of the circle denotes in which of the classes the enrichment occurs and the area of circle denotes the normalized enrichment score. The intensity of the colors shows the limit of significance of the enrichment or false discovery rate. Samples used in this analysis are listed in supplemental Table 3a.
Affymetrix gene-chip processing and analysis
Total RNA was purified from sorted subpopulations from peripheral blood and lesion specimens according to the Arcturus PicoPure RNA Isolation kit protocol (Applied Biosystems). RNA quality was verified using the Pico Chip at the Baylor University College of Medicine Microarray Facility. cDNA amplification was performed using the Ovation Pico WTA V2 system according to the manufacturer’s protocol (Nugen, San Carlos, CA). Fragmented and biotinylated cDNA was hybridized to GeneChip Human Transcriptome Array 2.0 according to the manufacturer’s procedures (Affymetrix, Waltham, MA). Raw data from all samples were normalized using the SST-RMA algorithm implemented in the Affymetrix Expression Console.
A 1-way analysis of variance was used to compare LCH CD1a+CD207+ DCs to healthy control blood CD1c+ mDCs. DEGs were identified using the Transcriptome Analysis Console 4.0 with false discovery rate controlled at 0.05 using the BH method and a fold change >2. All samples that were used in the analysis are listed in supplemental Table 3a. Among the DEGs, 2190 of them were differentially expressed between the 2 populations. A heatmap was generated showing the 50 genes with highest significant relative expression in LCH CD1a+CD207+ DCs (supplemental Figure 2).
A paired 1-way repeated measure analysis of variance was used to compare paired samples of CD1a+CD207high, CD1a+CD207low, CD1a+CD207− cells. Significance was determined using the false discovery rate controlled at 0.05 using the BH method. The samples used in analysis are listed in supplemental Table 3a.
A principal component analysis (PCA) was performed using the samples listed in supplemental Table 3a with R statistical software. The ggbiplot package was used to draw a normal data ellipse for each group.
Quantitative real-time PCR
Total RNA was isolated, and cDNA was generated as described in “Affymetrix gene chip processing and analysis.” Quantitative real-time polymerase chain reactions (PCRs) were performed with TaqMan gene-expression assays (Applied Biosystems) and the PrimePCR probe assay (Bio-Rad Laboratories, Hercules, CA) on a CFX96 real-time system (Bio-Rad Laboratories) according to the manufacturer’s protocol. TaqMan probe sets used were CD1a (HS00381754_g1), CD1c (HS00233509_m1), CD207 (Hs00210451_m1), CD141 (HS00264920_s1), CLEC9A (HS001651638_m1), HLA-DQA2 (Hs00607448_gH), HLA-DQB2 (Hs00794552_m1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; HS02758991_g1). Prime PCR probe sets were GAPDH (qHsaCEP0041396) and HLA-DQA2 (qHsaCIP0040206). All data were normalized to GAPDH expression. Statistical analyses were performed with Prism 7.0 (GraphPad Software, La Jolla, CA). All samples used in this analysis are listed in supplemental Table 3b.
Results
Transcriptome of LCH lesion CD1a+CD207+ DCs is most closely related to blood CD1c+ mDCs
To evaluate the relatedness of transcriptome of LCH lesion pathogenic DCs to potential precursor populations, we first compared full gene-expression profiles of the lesion CD1a+CD207+ DCs to gene signatures generated from human myeloid cells (supplemental Table 3a). All of these myeloid cells including CD14+ classical monocytes, CD16+ nonclassical monocytes, CD14+ DCs (also referred as CD14+ monocyte-derived macrophages), resident macrophages, epidermal LCs, CD1c+ mDCs, and CD141+ mDCs have been previously used in human and mouse DC ontogeny studies.24,39 According to the CMAP analysis, the CD1c+ mDC gene signature had the highest enrichment in the gene-expression profile of LCH lesion CD1a+CD207+ DCs, followed by CD141+ mDCs and LCs, then CD14+ DCs and CD14+ monocytes, with least enrichment for the gene signatures of macrophages and CD16+ monocytes (Figure 2A). Analyzing these data with a complementary approach, we also compared gene-expression profiles of LCH lesion CD1a+CD207+ DCs to gene-expression profiles from other cell types, including human peripheral blood and skin DCs, monocytes, and macrophages obtained from microarray data sets using the BubbleGUM platform (supplemental Table 3a).41 Consistent with CMAP analysis, the CD1c+ mDC gene signature had the highest degree of enrichment in LCH lesion CD1a+CD207+ DC gene set compared with gene-expression profiles from other DCs and macrophage and monocyte subsets (Figure 2B). Collectively, these data revealed that the transcriptome of lesion CD1a+CD207+ DCs in this series was most closely related to that of CD1c+ mDCs.

Transcriptome of LCH lesion CD1a+CD207+cells is most closely related to blood CD1c+mDCs. (A) CMAP analysis score for LCH CD1a+CD207+ DCs (n = 6) against healthy donor DCs/monocytes/macrophages after removal of tissue-specific genes. Each symbol represents an LCH lesion specimen. All samples used in the study are listed in supplemental Table 3a. A 1000 permutation test among gene signatures was performed on each enrichment score to determine the significance. CMAP scores for LCH CD1a+CD207+ DCs with all other human myeloid subsets were significant at P < .0001. The CMAP scores indicate the relative “closeness” of LCH CD1a+CD207+ DCs to myeloid subsets. LCH CD1a+CD207+ DCs show the highest CMAP scores with CD1c+ mDCs, followed by CD141+ mDCs and epidermal LCs. (B) Gene signatures of monocyte/DC/macrophage populations were used to compare with the gene-expression data set from LCH lesion CD1a+CD207+ DCs (n = 6) against the gene-expression data set from human myeloid subsets using BubbleGUM. Gene-expression profiles were obtained from FACS-sorted CD1a+CD207+ DCs from LCH lesions as well as DC/monocyte populations from healthy donor (HD) skin and peripheral blood specimens. Illumina Human HT-12 V4.0 was used for this study. All samples used in the study are listed in supplemental Table 3a. Blue bubbles represent similarity to the LCH CD1a+CD207+ transcriptome; red bubbles represent similarity to the comparison transcriptome. The bubble area corresponds to the GSEA normalized enrichment score (NES); the intensity of the color corresponds to the statistical significance of the enrichment. The larger and darker in color the bubble becomes, the more significant the enrichment of the gene signature becomes in that particular class. LCH CD1a+CD207+ DCs consistently show the highest enrichment (blue) with the CD1c+ mDC gene signature when compared with all signatures from other cell types. (C) Unsupervised cluster analysis of LCH lesion CD1a+CD207+ DCs and healthy donor blood DC/monocyte subsets demonstrates the relationship between LCH lesion CD1a+CD207+ DCs and healthy donor blood CD1c+ mDCs. Gene-expression profiles were obtained from FACS-sorted CD1a+CD207+ DCs from LCH lesions as well as DC/monocyte populations from healthy donors skin and peripheral blood specimens. Illumina Human HT-12 V4.0 was used for this study. All samples used in the study are listed in supplemental Table 3a. FDR, false discovery rate; NS, not significant.
Transcriptome of LCH lesion CD1a+CD207+cells is most closely related to blood CD1c+mDCs. (A) CMAP analysis score for LCH CD1a+CD207+ DCs (n = 6) against healthy donor DCs/monocytes/macrophages after removal of tissue-specific genes. Each symbol represents an LCH lesion specimen. All samples used in the study are listed in supplemental Table 3a. A 1000 permutation test among gene signatures was performed on each enrichment score to determine the significance. CMAP scores for LCH CD1a+CD207+ DCs with all other human myeloid subsets were significant at P < .0001. The CMAP scores indicate the relative “closeness” of LCH CD1a+CD207+ DCs to myeloid subsets. LCH CD1a+CD207+ DCs show the highest CMAP scores with CD1c+ mDCs, followed by CD141+ mDCs and epidermal LCs. (B) Gene signatures of monocyte/DC/macrophage populations were used to compare with the gene-expression data set from LCH lesion CD1a+CD207+ DCs (n = 6) against the gene-expression data set from human myeloid subsets using BubbleGUM. Gene-expression profiles were obtained from FACS-sorted CD1a+CD207+ DCs from LCH lesions as well as DC/monocyte populations from healthy donor (HD) skin and peripheral blood specimens. Illumina Human HT-12 V4.0 was used for this study. All samples used in the study are listed in supplemental Table 3a. Blue bubbles represent similarity to the LCH CD1a+CD207+ transcriptome; red bubbles represent similarity to the comparison transcriptome. The bubble area corresponds to the GSEA normalized enrichment score (NES); the intensity of the color corresponds to the statistical significance of the enrichment. The larger and darker in color the bubble becomes, the more significant the enrichment of the gene signature becomes in that particular class. LCH CD1a+CD207+ DCs consistently show the highest enrichment (blue) with the CD1c+ mDC gene signature when compared with all signatures from other cell types. (C) Unsupervised cluster analysis of LCH lesion CD1a+CD207+ DCs and healthy donor blood DC/monocyte subsets demonstrates the relationship between LCH lesion CD1a+CD207+ DCs and healthy donor blood CD1c+ mDCs. Gene-expression profiles were obtained from FACS-sorted CD1a+CD207+ DCs from LCH lesions as well as DC/monocyte populations from healthy donors skin and peripheral blood specimens. Illumina Human HT-12 V4.0 was used for this study. All samples used in the study are listed in supplemental Table 3a. FDR, false discovery rate; NS, not significant.
To further investigate whether LCH lesion CD1a+CD207+ cells are biologically related to less mature CD1c+ mDCs (blood CD1c+ mDCs) or more differentiated CD1c+ mDCs (tissue CD1c+ mDCs), we compared transcriptomes from LCH CD1a+CD207+ DCs to blood CD1c+ mDCs and skin CD1c+ mDCs along with CD14+ monocytes and CD14+ DCs through unsupervised hierarchical clustering. Interestingly, LCH lesion CD1a+CD207+ DCs clustered more closely to the blood CD1c+ mDCs than to the skin CD1c+ mDCs (Figure 2C).
Transcriptome analysis of LCH lesion cell populations
Although LCH lesion pathogenic cells are generally described as CD1a+CD207+, flow cytometry analysis of LCH lesions demonstrate some heterogeneity of the CD207 expression level variable relative abundance of CD1a+ subpopulations (Figure 3A; supplemental Table 5). LCH lesion CD1a+ subpopulations including CD1a+CD207high cells, CD1a+CD207low cells, and CD1a+CD207− cells were analyzed along with CD3+ lymphocytes from LCH lesions with respect to global gene expression (Figure 3A-B; supplemental Figure 4). PCA of gene-expression profiles from all subpopulations analyzed showed that CD1a+CD207−, CD1a+CD207high, and CD1a+CD207low subpopulations were tightly clustered relative to the CD3+ population (Figure 3C). Comparison of global gene-expression profiles of these 3 LCH lesion CD1a+ subpopulations did not identify any significantly differentially expressed genes after controlling for multiple comparisons. However, specific analysis of CD207 expression identified significantly increased expression from CD1a+CD207− through CD1a+CD207high cells with consistent levels of genes expressing CD1a, CD1c, and TNFα (supplemental Figure 1).
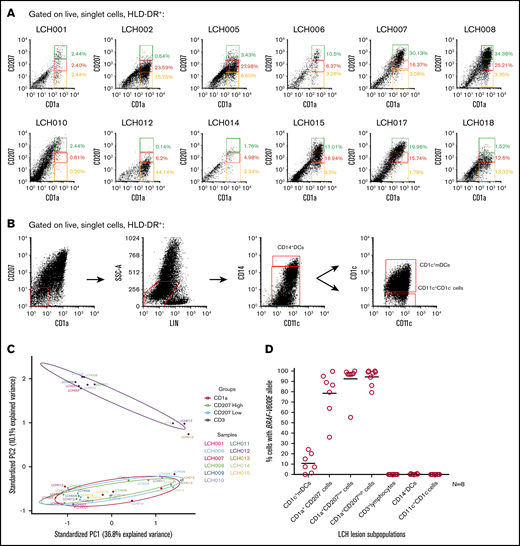
BRAFV600E+cells localize to LCH lesion CD1c+mDCs, CD1a+CD207−, CD1a+CD207low, and CD1a+CD207highsubpopulations. (A) Dot plots showing identification of 3 LCH subpopulations within HLA-DR+ and CD1a+ fractions from 12 LCH lesions: CD1a+CD207− (yellow gate), CD1a+CD207low (red gate), and CD1a+CD207high cells (green gate). (B) Flow cytometry of LCH lesion specimens with representative dot plots showing identification of 3 subpopulations within HLA-DR+, CD45+, CD207−CD1a−, and LIN− fractions from LCH lesions: CD14+ DCs, CD1c+ mDCs, and CD11c+CD1c− cells. (C) PCA of global transcriptome data from LCH lesion subpopulations (LCH lesion CD3+ lymphocytes, CD1a+CD207− cells, CD1a+CD207low cells and CD1a+CD207high cells) demonstrates clustering of all the CD1a+ populations and also clustering of the CD3+ populations. All samples used in the study are listed in supplemental Table 3a. (D) Lesions from LCH patients (N = 8) (supplemental Table 3b) were FACS-purified according to gating strategies shown in Figure 3A-B and supplemental Figure 4. Genomic DNA was extracted and amplified, and then the BRAFV600E allele was quantified by quantitative PCR (qPCR) as described in Berres et al.3 BRAFV600E was highly enriched in all CD1a+ populations, and was also detected in CD1c+ mDCs, but not in other LCH lesion populations. Technical duplicates were used in this experiment. CD207 and CD1a expression in LCH lesion subpopulations was further determined and shown in supplemental Figure 3. PC1, first principal component; PC2, second principal component.
BRAFV600E+cells localize to LCH lesion CD1c+mDCs, CD1a+CD207−, CD1a+CD207low, and CD1a+CD207highsubpopulations. (A) Dot plots showing identification of 3 LCH subpopulations within HLA-DR+ and CD1a+ fractions from 12 LCH lesions: CD1a+CD207− (yellow gate), CD1a+CD207low (red gate), and CD1a+CD207high cells (green gate). (B) Flow cytometry of LCH lesion specimens with representative dot plots showing identification of 3 subpopulations within HLA-DR+, CD45+, CD207−CD1a−, and LIN− fractions from LCH lesions: CD14+ DCs, CD1c+ mDCs, and CD11c+CD1c− cells. (C) PCA of global transcriptome data from LCH lesion subpopulations (LCH lesion CD3+ lymphocytes, CD1a+CD207− cells, CD1a+CD207low cells and CD1a+CD207high cells) demonstrates clustering of all the CD1a+ populations and also clustering of the CD3+ populations. All samples used in the study are listed in supplemental Table 3a. (D) Lesions from LCH patients (N = 8) (supplemental Table 3b) were FACS-purified according to gating strategies shown in Figure 3A-B and supplemental Figure 4. Genomic DNA was extracted and amplified, and then the BRAFV600E allele was quantified by quantitative PCR (qPCR) as described in Berres et al.3 BRAFV600E was highly enriched in all CD1a+ populations, and was also detected in CD1c+ mDCs, but not in other LCH lesion populations. Technical duplicates were used in this experiment. CD207 and CD1a expression in LCH lesion subpopulations was further determined and shown in supplemental Figure 3. PC1, first principal component; PC2, second principal component.
BRAFV600E localizes to CD1a+CD207+ cells, CD1a+CD207− cells, and CD1c+ mDCs within the LCH lesion
Somatic BRAFV600E mutation alleles have been previously demonstrated to be enriched in LCH lesion CD1a+CD207+ DCs.3,43 Assuming BRAFV600E to be a driver mutation and cells to be clonal, candidate precursor and pathogenic cells would be expected to share this mutation. We therefore analyzed BRAFV600E allele burden in LCH lesion subtypes and found that, in addition to CD1a+CD207high, CD1a+CD207low, and CD1a+CD207− cells, the BRAFV600E allele could also be identified in CD1c+ mDCs, but was not detected in other LCH lesion subpopulations, including CD11c+CD1c− cells that contain myeloid precursors, monocytes, and CD141+ mDCs (Figure 3D).
HLA-DQB2 expression is restricted to LCH lesion CD1a+ DC subpopulations, LCH lesion CD1c+ mDCs, and blood CD1c+ mDCs in patients with active high-risk LCH
These gene-expression and mutation-mapping studies identified CD1c+ mDCs as transcriptionally and mutationally similar to LCH lesion CD1a+CD207+ cells. To identify potential biomarkers specific to differentiating DCs, we compared the gene-expression profiles between healthy donor blood CD1c+ mDCs and LCH lesion CD1a+CD207+ DCs. This analysis identified differential expression of known LCH-related genes including CD1a, CD1c, CD207, and TNFα, which were much more highly expressed in LCH lesion CD1a+CD207+ DCs relative to blood CD1c+ mDCs from healthy donors (Figure 4A; supplemental Figure 2). An unexpected finding from this experiment was significantly increased expression of HLA-DQA2 and HLA-DQB2 in LCH lesion CD1a+CD207+ cells (Figure 4A). These genes were not previously associated with LCH, but have been previously reported to be specific to human epidermal LCs.44 HLA-DQA2 and HLA-DQB2 expression was further assessed across LCH lesion subpopulations (Figure 4B; supplemental Figure 5), with purity of all sorted LCH subpopulations confirmed by real-time PCR (supplemental Figure 3A-B). Where HLA-DQA2 was expressed in many subpopulations in our study, HLA-DQB2 expression was specific to the LCH lesion subpopulations that harbored BRAFV600E mutations (Figure 4B; supplemental Figure 5). Further flow cytometry analysis of LCH lesions identified surface expression of HLA-DQB2 on CD1c+ mDCs, CD1a+CD207− cells, CD1a+CD207low cells, and CD1a+CD207high cells, but not in other lesion myeloid and lymphoid subpopulations (Figure 4C; supplemental Figure 6). Gene-expression analysis found HLA-DQB2 in CD1a+CD207+ cells from low-risk and high-risk lesions, with no statistical difference in expression between risk groups (supplemental Figure 7).
![HLA-DQB2 is specifically expressed in LCH CD1c+mDCs, CD1a+CD207−, CD1a+CD207low, and CD1a+CD207highsubpopulations. (A) Scatter plots illustrate expression of 6 genes (CD1a, CD1c, CD207, TNFα, HLADQA2, and HLADQB2) among the top 50 most overexpressed genes in LCH CD1a+CD207+ DCs relative to healthy donor blood CD1c+ mDCs. Gene-expression profiles were obtained from FACS-sorted LCH lesion CD1a+CD207+ DCs (n = 11) and blood CD1c+ mDCs (n = 3) from healthy donors. Affymetrix Human Transcriptome Array 2.0 Platform was used for this study. All samples used in the study are listed in supplemental Table 3a. (B) Control skin specimens (n = 4), peripheral blood samples (n = 4) from healthy donors, and lesion from LCH patients (n = 6) were FACS-purified. RNA was extracted, cDNA was amplified and then the HLA-DQB2 expression was determined by qPCR (normalized to GAPDH messenger RNA [mRNA] expression). HLA-DQB2 expression was detected in lesion CD1a+ populations as well as CD1c+ mDCs, but not in other lineages, consistent with BRAFV600E distribution. Epidermal LCs were used as a positive control. Technical duplicates were used. LCH samples used in the study are listed in supplemental Table 3b. (C) Overlay histograms of HLA-DQB2 surface expression in 6 LCH lesion subpopulations (colored) compared with control (gray) demonstrates increasing surface expression of HLA-DQB2 from lesion CD1c+ mDCs through CD1a+CD207+ populations.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/1/10.1182_bloodadvances.2019000488/3/m_advancesadv2019000488f4.png?Expires=1765888852&Signature=roopAmOk4NaVCFF6u0NhwbuAj~fiETaH9Z5C3TwaFdHe3BV9k-ln6drsgdBXnP2hPJnWtZYcYPvL1h~4J6TelwmmHez7FOkirvfJt7TgoVTJSSbe-j6nYqevE7io3k7VnkHcoCmVhZDZGsXVH5WlZUv6fYvS18gojEm5kJVrrfU049Tr9ymlXSxBZF60XEwF5~5IKZ9a8aIJQSAm0aJmn7gTuXahfPn6zN37~nKs2EK8K8YL44tXgI3uzqP-s8E7HMUzERKEqOOai--aHQwV9oQaor08YlMLkqaDNEQJo4ce67Q4IUg7AxBx-2sjGWCZEV25ApdIki6j88ojjS3vpg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HLA-DQB2 is specifically expressed in LCH CD1c+mDCs, CD1a+CD207−, CD1a+CD207low, and CD1a+CD207highsubpopulations. (A) Scatter plots illustrate expression of 6 genes (CD1a, CD1c, CD207, TNFα, HLADQA2, and HLADQB2) among the top 50 most overexpressed genes in LCH CD1a+CD207+ DCs relative to healthy donor blood CD1c+ mDCs. Gene-expression profiles were obtained from FACS-sorted LCH lesion CD1a+CD207+ DCs (n = 11) and blood CD1c+ mDCs (n = 3) from healthy donors. Affymetrix Human Transcriptome Array 2.0 Platform was used for this study. All samples used in the study are listed in supplemental Table 3a. (B) Control skin specimens (n = 4), peripheral blood samples (n = 4) from healthy donors, and lesion from LCH patients (n = 6) were FACS-purified. RNA was extracted, cDNA was amplified and then the HLA-DQB2 expression was determined by qPCR (normalized to GAPDH messenger RNA [mRNA] expression). HLA-DQB2 expression was detected in lesion CD1a+ populations as well as CD1c+ mDCs, but not in other lineages, consistent with BRAFV600E distribution. Epidermal LCs were used as a positive control. Technical duplicates were used. LCH samples used in the study are listed in supplemental Table 3b. (C) Overlay histograms of HLA-DQB2 surface expression in 6 LCH lesion subpopulations (colored) compared with control (gray) demonstrates increasing surface expression of HLA-DQB2 from lesion CD1c+ mDCs through CD1a+CD207+ populations.
HLA-DQB2 is specifically expressed in LCH CD1c+mDCs, CD1a+CD207−, CD1a+CD207low, and CD1a+CD207highsubpopulations. (A) Scatter plots illustrate expression of 6 genes (CD1a, CD1c, CD207, TNFα, HLADQA2, and HLADQB2) among the top 50 most overexpressed genes in LCH CD1a+CD207+ DCs relative to healthy donor blood CD1c+ mDCs. Gene-expression profiles were obtained from FACS-sorted LCH lesion CD1a+CD207+ DCs (n = 11) and blood CD1c+ mDCs (n = 3) from healthy donors. Affymetrix Human Transcriptome Array 2.0 Platform was used for this study. All samples used in the study are listed in supplemental Table 3a. (B) Control skin specimens (n = 4), peripheral blood samples (n = 4) from healthy donors, and lesion from LCH patients (n = 6) were FACS-purified. RNA was extracted, cDNA was amplified and then the HLA-DQB2 expression was determined by qPCR (normalized to GAPDH messenger RNA [mRNA] expression). HLA-DQB2 expression was detected in lesion CD1a+ populations as well as CD1c+ mDCs, but not in other lineages, consistent with BRAFV600E distribution. Epidermal LCs were used as a positive control. Technical duplicates were used. LCH samples used in the study are listed in supplemental Table 3b. (C) Overlay histograms of HLA-DQB2 surface expression in 6 LCH lesion subpopulations (colored) compared with control (gray) demonstrates increasing surface expression of HLA-DQB2 from lesion CD1c+ mDCs through CD1a+CD207+ populations.
The BRAFV600E mutation may be detected at low levels (generally <0.1%) in PBMCs) of patients with active BRAFV600E+ lesions characterized as high risk (liver, spleen, and/or bone marrow involvement).3 Using real-time PCR to characterize HLA-DQB2 expression in PBMCs of patients with active BRAFV600E+ LCH lesions, HLA-DQB2 expression was identified in PBMCs of high-risk LCH, but not in PBMCs from both low-risk multisystem and low-risk single system patients, correlating with identification of detectable BRAFV600E allele (Figure 5A-B). PBMCs were then analyzed by flow cytometry for HLA-DQB2 surface expression. Interestingly, HLA-DQB2 expression was only identified in high-risk LCH patients (but not in low-risk patients or healthy controls) (Figure 5C; supplemental Figure 8), and it was localized to CD1c+ mDCs (3.3% to 23.7% of total peripheral blood CD1c+ mDCs) (Figure 5C). Although the BRAFV600E allele was identified across multiple peripheral blood cell subpopulations, it was highly enriched in CD1c+HLA-DQB2+ mDCs (Figure 5D-E).3,17
![HLA-DQB2 expression is identified on blood BRAF-V600E+CD1c+mDCs in patients with high-risk (HR) LCH. (A) Genomic DNA was isolated from peripheral blood specimens from LCH patients (N = 11; n = 3 for high-risk multisystem, n = 3 for low-risk [LR] multisystem, n = 5 for low-risk single lesion) with BRAFV600E+ lesions and healthy donors (N = 11). The percentage of circulating cells with BRAFV600E allele was determined by qPCR.3 As expected from previous studies, BRAFV600E expression was detected at low levels in PBMCs from patients with BRAFV600E+ lesions and active high-risk LCH, and was absent from PBMCs from patients with low-risk LCH and healthy donors (HD).3 Technical duplicates were used in this experiment. LCH patients’ PBMC samples used in the study are listed in supplemental Table 1b. Blue dot, Patient LCH 0019; red dot, patient LCH 0020; green dot, patient LCH 0021. (B) RNA from peripheral blood specimens from the same set of LCH patients as in panel A was extracted and cDNA was amplified, and then the HLA-DQB2 expression was determined by qPCR (normalized to GAPDH mRNA expression). HLA-DQB2 expression was specifically detected in PBMCs from the same patients with detectable BRAFV600E+ PBMCs. Technical duplicates were used in this experiment. Blue dot, Patient LCH 0019; red dot, patient LCH 0020; green dot, patient LCH 0021). (C) Representative dot plots showing identification of CD1c+ mDCs (green gate) within HLA-DR+ cells from PBMCs of an HR LCH patient. Overlay histograms show HLA-DQB2 expression in LCH lesion CD1c+ mDCs (green) compared with control (gray). HLA-DQB2 expression was detectable on some CD1c+ mDCs. Representative dot plots showing identification of CD1c+ mDCs (green gate) from PBMCs of a healthy donor were illustrated in supplemental Figure 8. No HLA-DQB2 expression was detectable on CD1c+ mDCs from PBMCs of a healthy donor. (D) Genomic DNA from unsorted and sorted cells from PBMCs of high-risk LCH patients (N = 3) with BRAFV600E+ lesions was isolated and amplified, and the percentage of cells with BRAFV600E allele was determined by qPCR. As demonstrated in previous studies, many lineages have the potential to carry the BRAFV600E mutation.3 Technical duplicates were used in this experiment. Blue dot, Patient LCH 0019; red dot, patient LCH 0020; green dot, patient LCH 0021. (E) Genomic DNA from CD1c+ mDCs (HLA-DQB2- and HLA-DQB2+) from PBMCs of high-risk LCH patients (N = 3) was isolated and amplified, and the percentage of cells with BRAFV600E allele was determined by qPCR. BRAFV600E was highly enriched in the HLA-DQB2+CD1c+ mDC population. Technical duplicates were used in this experiment. Blue dot, Patient LCH 0019; red dot, patient LCH 0020; green dot, patient LCH 0021.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/1/10.1182_bloodadvances.2019000488/3/m_advancesadv2019000488f5.png?Expires=1765888852&Signature=wDKwGqkMDJHBa6yAPvJk354CwZe9eiX~xYyB3zXVbfsfy8sHdUh-yWUaqxY7pALEsFuNGCbVJ38OF5O7J91fgZUtM~WfG~BFsGvWKF~LRmGzvg2tiKL2COFST0yHldTw48BvUcOe8W~~NcXUQ-GnaRYm~T6PS4NJREFvgDQn7lAaAEMtH7rWWZFNJ9vIZUrI6QwKORnkH-rPmNlj0MtfDfuDBLk9qYyANHQ2b66AoFv9eGZ5p0S8lvrHyK-QacfrvuuT481NvtGoOTNFRKyi9x4pjfz~7zwmgbR3NkCb1gsJsmSThHxPnToWhC39n2VL6-T2paUh2iEfGGClc9xpuA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HLA-DQB2 expression is identified on blood BRAF-V600E+CD1c+mDCs in patients with high-risk (HR) LCH. (A) Genomic DNA was isolated from peripheral blood specimens from LCH patients (N = 11; n = 3 for high-risk multisystem, n = 3 for low-risk [LR] multisystem, n = 5 for low-risk single lesion) with BRAFV600E+ lesions and healthy donors (N = 11). The percentage of circulating cells with BRAFV600E allele was determined by qPCR.3 As expected from previous studies, BRAFV600E expression was detected at low levels in PBMCs from patients with BRAFV600E+ lesions and active high-risk LCH, and was absent from PBMCs from patients with low-risk LCH and healthy donors (HD).3 Technical duplicates were used in this experiment. LCH patients’ PBMC samples used in the study are listed in supplemental Table 1b. Blue dot, Patient LCH 0019; red dot, patient LCH 0020; green dot, patient LCH 0021. (B) RNA from peripheral blood specimens from the same set of LCH patients as in panel A was extracted and cDNA was amplified, and then the HLA-DQB2 expression was determined by qPCR (normalized to GAPDH mRNA expression). HLA-DQB2 expression was specifically detected in PBMCs from the same patients with detectable BRAFV600E+ PBMCs. Technical duplicates were used in this experiment. Blue dot, Patient LCH 0019; red dot, patient LCH 0020; green dot, patient LCH 0021). (C) Representative dot plots showing identification of CD1c+ mDCs (green gate) within HLA-DR+ cells from PBMCs of an HR LCH patient. Overlay histograms show HLA-DQB2 expression in LCH lesion CD1c+ mDCs (green) compared with control (gray). HLA-DQB2 expression was detectable on some CD1c+ mDCs. Representative dot plots showing identification of CD1c+ mDCs (green gate) from PBMCs of a healthy donor were illustrated in supplemental Figure 8. No HLA-DQB2 expression was detectable on CD1c+ mDCs from PBMCs of a healthy donor. (D) Genomic DNA from unsorted and sorted cells from PBMCs of high-risk LCH patients (N = 3) with BRAFV600E+ lesions was isolated and amplified, and the percentage of cells with BRAFV600E allele was determined by qPCR. As demonstrated in previous studies, many lineages have the potential to carry the BRAFV600E mutation.3 Technical duplicates were used in this experiment. Blue dot, Patient LCH 0019; red dot, patient LCH 0020; green dot, patient LCH 0021. (E) Genomic DNA from CD1c+ mDCs (HLA-DQB2- and HLA-DQB2+) from PBMCs of high-risk LCH patients (N = 3) was isolated and amplified, and the percentage of cells with BRAFV600E allele was determined by qPCR. BRAFV600E was highly enriched in the HLA-DQB2+CD1c+ mDC population. Technical duplicates were used in this experiment. Blue dot, Patient LCH 0019; red dot, patient LCH 0020; green dot, patient LCH 0021.
HLA-DQB2 expression is identified on blood BRAF-V600E+CD1c+mDCs in patients with high-risk (HR) LCH. (A) Genomic DNA was isolated from peripheral blood specimens from LCH patients (N = 11; n = 3 for high-risk multisystem, n = 3 for low-risk [LR] multisystem, n = 5 for low-risk single lesion) with BRAFV600E+ lesions and healthy donors (N = 11). The percentage of circulating cells with BRAFV600E allele was determined by qPCR.3 As expected from previous studies, BRAFV600E expression was detected at low levels in PBMCs from patients with BRAFV600E+ lesions and active high-risk LCH, and was absent from PBMCs from patients with low-risk LCH and healthy donors (HD).3 Technical duplicates were used in this experiment. LCH patients’ PBMC samples used in the study are listed in supplemental Table 1b. Blue dot, Patient LCH 0019; red dot, patient LCH 0020; green dot, patient LCH 0021. (B) RNA from peripheral blood specimens from the same set of LCH patients as in panel A was extracted and cDNA was amplified, and then the HLA-DQB2 expression was determined by qPCR (normalized to GAPDH mRNA expression). HLA-DQB2 expression was specifically detected in PBMCs from the same patients with detectable BRAFV600E+ PBMCs. Technical duplicates were used in this experiment. Blue dot, Patient LCH 0019; red dot, patient LCH 0020; green dot, patient LCH 0021). (C) Representative dot plots showing identification of CD1c+ mDCs (green gate) within HLA-DR+ cells from PBMCs of an HR LCH patient. Overlay histograms show HLA-DQB2 expression in LCH lesion CD1c+ mDCs (green) compared with control (gray). HLA-DQB2 expression was detectable on some CD1c+ mDCs. Representative dot plots showing identification of CD1c+ mDCs (green gate) from PBMCs of a healthy donor were illustrated in supplemental Figure 8. No HLA-DQB2 expression was detectable on CD1c+ mDCs from PBMCs of a healthy donor. (D) Genomic DNA from unsorted and sorted cells from PBMCs of high-risk LCH patients (N = 3) with BRAFV600E+ lesions was isolated and amplified, and the percentage of cells with BRAFV600E allele was determined by qPCR. As demonstrated in previous studies, many lineages have the potential to carry the BRAFV600E mutation.3 Technical duplicates were used in this experiment. Blue dot, Patient LCH 0019; red dot, patient LCH 0020; green dot, patient LCH 0021. (E) Genomic DNA from CD1c+ mDCs (HLA-DQB2- and HLA-DQB2+) from PBMCs of high-risk LCH patients (N = 3) was isolated and amplified, and the percentage of cells with BRAFV600E allele was determined by qPCR. BRAFV600E was highly enriched in the HLA-DQB2+CD1c+ mDC population. Technical duplicates were used in this experiment. Blue dot, Patient LCH 0019; red dot, patient LCH 0020; green dot, patient LCH 0021.
Discussion
According to the Misguided Myeloid DC Model, LCH DCs arise from hematopoietic stem cells or myeloid precursors with pathologically activated MAPK signaling, and the extent of disease depends on the stage of differentiation of the myeloid precursor in which the activating mutation arises.1,12 Analyses of peripheral blood from children and adults with LCH demonstrate that the BRAFV600E mutation may be carried by a range of myeloid and lymphoid cells.3,17 We hypothesize that LCH lesion CD1a+CD207+ DCs share a common pathway of terminal differentiation, though the pathway from precursor to CD1a+CD207+ DC remains undefined. In this study, we aimed to identify potential origin(s) of LCH lesion CD1a+CD207+ DCs.
Given the ability to differentiate into langerin+ (CD207+) cells in vitro, both CD1c+ mDCs and CD14+ monocytes are potential candidates to give rise to LCH CD1a+CD207+ DCs.14,15,17,31,45 To determine the differentiation pathway(s) of LCH CD1a+CD207+ DCs, we first compared the gene-expression profiles of CD1a+CD207+ DCs to gene signatures from human myeloid cell lineages. LCH CD1a+CD207+ cells were most highly enriched with CD1c+ mDC signature, with relatively lower enrichment with the CD14+monocyte signature using both CMAP and BubbleGum analysis platforms (Figure 2A-B). Furthermore, unsupervised clustering of transcriptomic data demonstrated that LCH CD1a+CD207+ DCs were more related to blood CD1c+ mDCs than to tissue CD1c+ mDCs, blood CD14+ monocytes, or tissue CD14+ DCs (Figure 2C). Analysis of BRAFV600E allele burden in LCH lesion cells identified the mutation in CD1a+CD207− cells, CD1a+CD207low cells, and CD1a+CD207high cells as well as in CD1c+ mDCs, but not in lesion CD14+ DCs (Figure 3D). These transcriptome analysis and mutation-tracing experiments are consistent with blood CD1c+ mDC as a potential precursor to the LCH lesion CD1a+CD207+ DC.
It is therefore plausible that blood CD1c+ mDC could migrate to the lesion, become tissue CD1c+ mDC, and then acquire CD1a and CD207 expression (Figure 6). To test changes in gene expression along such a pathway of differentiation, we compared gene-expression profiles of LCH lesion CD1a+CD207+ cells and healthy control blood CD1c+ mDCs. In addition to some expected differences in gene expression (CD207, CD1a, TNF, MMP9), an unexpected finding was remarkably increased expression of HLA-DQA2 and HLA-DQB2 in LCH lesion CD1a+CD207+ DCs (Figure 4A; supplemental Figure 2). These HLA class II (HLA-II) genes were previously reported with physiologic expression specific to epidermal LCs.44 In our study, we found HLA-DQA2 expression in multiple myeloid populations in LCH lesions (supplemental Figure 5). However, HLA-DQB2 expression was specific to CD1c+ mDCs, CD1a+CD207− cells, and CD1a+CD207+ cells (Figure 4B-C), cells with the common feature of also harboring the BRAFV600E allele (Figure 3D).
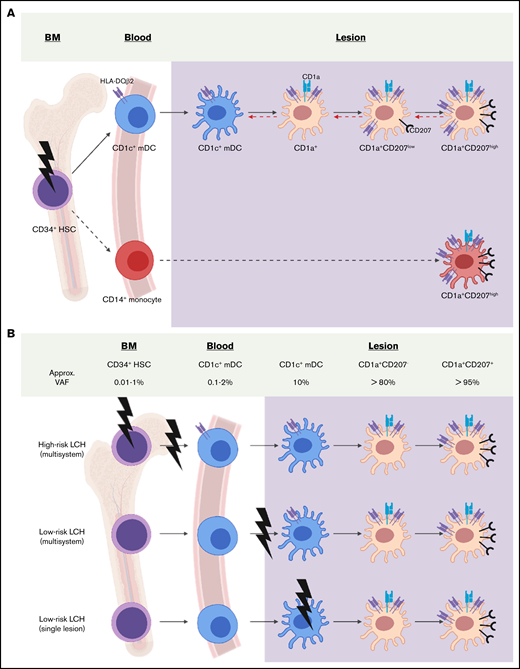
Models of LCH DC differentiation. (A) Previous studies support a model of LCH ontogeny where activating MAPK pathway gene mutation (bolt) in hematopoietic stem and precursor cells (gray) drive LCH lesion formation.3,17,47 This study adds to previous reports by demonstrating HLA-DQB2 (purple surface icon) expression on blood CD1c+ mDCs with BRAFV600E+ in patients with high-risk LCH. Within LCH lesions, the presence of BRAFV600E and HLADQB2 in CD1c+ mDCs, CD1a+CD207− cells, CD1a+CD207low cells, and CD1a+CD207high cells is consistent with differentiation of the CD1c+ mDC precursor from blood into lesion DCs. A logical pathway (solid black arrow) would lead to acquisition of CD1a, then CD207 expression; however, it is also possible that these populations may exist in equilibrium (dashed red arrow) at different stages of terminal differentiation based on environmental cues. Although data from this study support origin of LCH CD1a+CD207+ DCs from blood CD1c+ mDCs, it remains possible that some LCH DCs may arise from blood CD14+ monocytes, though lack of BRAFV600E+CD14+ cells in lesions would require these cells to rapidly differentiate (dashed black arrow) or represent a minor fraction of LCH lesion cells below the limits of detection. (B) This schema shows the Misguided Myeloid Differentiation Model for LCH ontogeny for different risk groups, augmented with data from this study.1 According to this model, the stage of differentiation in which myeloid cell acquires BRAFV600E mutation (or alternative activating MAPK gene mutations) determines the extent of LCH (high- or low-risk LCH). High-risk multisystem LCH arises from activating the MAPK gene mutation of hematopoietic stem/progenitor cells from bone marrow (BM); the low-risk multisystem LCH arises from somatic mutation of committed DC precursor cells in blood; and low-risk single system LCH arises from somatic mutation of more differentiated DC precursors from blood. Although the stage of differentiation in which myeloid cells acquire mutation defines LCH clinical manifestation, data from this study are consistent with blood-derived CD1c+ mDCs migrating to lesion sites and differentiating into pathologic CD1a+CD207+ LCH cells. VAF, variant allele frequency.
Models of LCH DC differentiation. (A) Previous studies support a model of LCH ontogeny where activating MAPK pathway gene mutation (bolt) in hematopoietic stem and precursor cells (gray) drive LCH lesion formation.3,17,47 This study adds to previous reports by demonstrating HLA-DQB2 (purple surface icon) expression on blood CD1c+ mDCs with BRAFV600E+ in patients with high-risk LCH. Within LCH lesions, the presence of BRAFV600E and HLADQB2 in CD1c+ mDCs, CD1a+CD207− cells, CD1a+CD207low cells, and CD1a+CD207high cells is consistent with differentiation of the CD1c+ mDC precursor from blood into lesion DCs. A logical pathway (solid black arrow) would lead to acquisition of CD1a, then CD207 expression; however, it is also possible that these populations may exist in equilibrium (dashed red arrow) at different stages of terminal differentiation based on environmental cues. Although data from this study support origin of LCH CD1a+CD207+ DCs from blood CD1c+ mDCs, it remains possible that some LCH DCs may arise from blood CD14+ monocytes, though lack of BRAFV600E+CD14+ cells in lesions would require these cells to rapidly differentiate (dashed black arrow) or represent a minor fraction of LCH lesion cells below the limits of detection. (B) This schema shows the Misguided Myeloid Differentiation Model for LCH ontogeny for different risk groups, augmented with data from this study.1 According to this model, the stage of differentiation in which myeloid cell acquires BRAFV600E mutation (or alternative activating MAPK gene mutations) determines the extent of LCH (high- or low-risk LCH). High-risk multisystem LCH arises from activating the MAPK gene mutation of hematopoietic stem/progenitor cells from bone marrow (BM); the low-risk multisystem LCH arises from somatic mutation of committed DC precursor cells in blood; and low-risk single system LCH arises from somatic mutation of more differentiated DC precursors from blood. Although the stage of differentiation in which myeloid cells acquire mutation defines LCH clinical manifestation, data from this study are consistent with blood-derived CD1c+ mDCs migrating to lesion sites and differentiating into pathologic CD1a+CD207+ LCH cells. VAF, variant allele frequency.
Given the expression pattern of HLA-DQB2 on potential LCH lesion CD1a+CD207+ DC precursors, we investigated peripheral blood populations for HLA-DQB2 expression. As in the study by Lenormand et al,44 HLA-DQB2 expression was absent from any peripheral blood populations from healthy donors. However, we did identify HLA-DQB2 expression in CD1c+ mDCs in peripheral blood of patients with high-risk LCH (Figure 5C). Despite the presence of BRAFV600E among multiple PBMC populations in patients with high-risk LCH (and BRAFV600E+ lesions), HLA-DQB2 localized to CD1c+ mDCs (Figure 5D-C). Furthermore, BRAFV600E alleles were enriched in CD1c+HLA-DQB2+ mDCs (Figure 5E). HLA-DQB2 mRNA and cell-surface expression may therefore represent novel biomarkers for circulating LCH precursors and pathologic LCH lesion DCs. HLA-DQB2 was demonstrated to stimulate T cells in vitro.44 It is plausible that, in addition to representing a clinically relevant biomarker, it may play a role in mediating inflammation in LCH lesions.
Although this study supports a model of CD1c+ mDCs as precursors for LCH CD1a+CD207+ DCs, it is possible that >1 cell of origin may exist. It has been shown previously that CD14+ monocytes have the capacity to become CD1a+CD207+ cells in response to notch ligands δ-like protein 1 (DLL1), DLL4, GMCSF, and TGFβ in vitro.14,15,17,31 Furthermore, a recent study showed that CD14+ monocytes could acquire gene signature with similarities to the LCH CD1a+CD207+ cell signature in the presence of Notch ligands JAG2 in vitro.45 It remains possible that CD14+monocytes could differentiate rapidly into LCH CD1a+CD207+ DCs once entering the lesion, or that a small population of CD14+ DCs that present in LCH lesions may not be captured by the experiments in this study. Notably, unlike a previous report,46 we did not detect any CD207+ cells in peripheral blood of either LCH patients (high or low risk) or healthy controls (supplemental Figure 9).
The findings from this study support an updated Misguided Myeloid DC Model of LCH pathogenesis in which blood CD1c+ mDCs with hyperactive ERK may have the capacity to migrate to the LCH lesion sites and differentiate into LCH CD1a+CD207+ DCs (Figure 6). If differentiation from CD1c+ mDCs to CD1a+CD207+ DCs is critical to LCH pathogenesis, then blocking chemokines or integrins that allow CD1c+ mDCs to enter tissues may represent a novel therapeutic strategy. Additionally, these findings suggest potential roles of MAPK activation in physiologic CD1c+ mDC differentiation.
The gene-expression data reported in this article have been deposited in the Gene Expression Omnibus database (accession numbers GSE122476, GSE122673, and GSE122674). Samples used in this analysis have been deposited in the Gene Expression Omnibus database (accession numbers GSE35457, GSE85305, GSE122476 [secure token: chexggcmpzyppcj], GSE122673 [secure token: shqfsiacpxkzlap], and GSE122674 (secure token: qtipmwosjjwjbex).
Data that support the findings of the study are available from corresponding author Carl E. Allen at ceallen@txch.org upon reasonable request.
Acknowledgments
The authors thank Linna Zhang and Elizabeth Pacheco (College of Medicine, Baylor University; Texas Children’s Cancer Center) for data management support, and Tatiana Goltsova and Amos S. Gaikwad from the Flow Cytometry Core Laboratory (Texas Children’s Hospital) for assistance with flow cytometry.
The TXCH Histiocytosis Program is supported by a research grant from the HistioCure Foundation. Additional support was received from National Institutes of Health, National Cancer Institute grants R01 CA154489 (M.M. and C.E.A.), CA154947 (M.M. and C.E.A.), and P50CA126752 (Specialized Program of Research Excellence in Lymphoma) (C.E.A.); the St. Baldrick’s Foundation Innovation grant (C.E.A.); the St. Baldrick’s Foundation Consortium grant supporting the North American Consortium for Histiocytosis (NACHO) (C.E.A.); the Leukemia & Lymphoma Society Translational Research Program (C.E.A.); Singapore Immunology Network core funding (F.G.); a Histiocytosis Association grant (F.G.); and Cancer Research UK grant C30484/A21025 (M.C.). F.G. is an EMBO Young Investigator Programme awardee. K.P.H.L. was supported by the Howard Hughes Medical Institute to the Baylor University College of Medicine Med into Grad Initiative; National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases T32 training grants in Hematology (5T32DK60445-13 and 5T32DK60445-14); and a National Institutes of Health, National Institute of General Medical Sciences T32 training grant in Translational Biology and Molecular Medicine (2T32GM88129-6). T.M.B. was supported by National Cancer Institute Ruth L. Kirschstein National Research Service Award, Individual Predoctoral Fellowship to Promote Diversity in Health-Related Research grant 5F31CA228360-02. O.S.E. was supported by National Institutes of Health, National Cancer Institute K12 grant 2K12CA090433-16.
Authorship
Contribution: K.P.H.L. and P.M. designed research, performed experiments, analyzed data, and wrote, reviewed, and edited the manuscript; M.P., K.D., H.L., and N.M. designed research, analyzed data, and reviewed and edited the manuscript; H.A., D.Z., T.M.B., O.S.E., R.C., A.S., and B.S. provided technical advice, performed experiments, reviewed clinical data, and reviewed and edited the manuscript; E.N., M.M., K.L.M., and T.-K.M. contributed to the study concept and experimental design and reviewed and edited the manuscript; and F.G., M.C., and C.E.A. conceived the study, supervised the project, and wrote, reviewed, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Carl E. Allen, Texas Children’s Hospital, Feigin Center, Suite 730.06, 1102 Bates St, Houston, TX 77030; e-mail: ceallen@txch.org; Matthew Collin, Human Dendritic Cell Laboratory, Institute of Cellular Medicine, Newcastle University, Framlington Pl, Newcastle upon Tyne NE2 4HH, United Kingdom; e-mail: matthew.collin@ncl.ac.uk; or Florent Ginhoux, Singapore Immunology Network (SIgN), Agency for Science, Technology and Research (A*STAR), Singapore 138648; e-mail: florent_ginhoux@immunol.a-star.edu.sg.

