

Key Points
Ovarian cancer organ-on-a-chip technology models platelet extravasation through the endothelium into tumors.
OvCa-Chip includes cocultures of cancer and vascular cells, and platelet flow revealing time-dependent events within tumor microenvironment.

Abstract
In ovarian cancer, platelet extravasation into the tumor and resulting metastasis is thought to be regulated mostly by the vascular endothelium. Because it is difficult to dissect complex underlying events in murine models, organ-on-a-chip methodology is applied to model vascular and platelet functions in ovarian cancer. This system (OvCa-Chip) consists of microfluidic chambers that are lined by human ovarian tumor cells interfaced with a 3-dimensional endothelialized lumen. Subsequent perfusion with human platelets within the device’s vascular endothelial compartment under microvascular shear conditions for 5 days uncovered organ-to-molecular–level contributions of the endothelium to triggering platelet extravasation into tumors. Further, analysis of effluents available from the device’s individual tumor and endothelial chambers revealed temporal dynamics of vascular disintegration caused by cancer cells, a differential increase in cytokine expression, and an alteration of barrier maintenance genes in endothelial cells. These events, when analyzed within the device over time, made the vascular tissue leaky and promoted platelet extravasation. Atorvastatin treatment of the endothelial cells within the OvCa-Chip revealed improved endothelial barrier function, reduction in inflammatory cytokines and, eventually, arrest of platelet extravasation. These data were validated through corresponding observations in patient-derived tumor samples. The OvCa-Chip provides a novel in vitro dissectible platform to model the mechanisms of the cancer-vascular-hematology nexus and the analyses of potential therapeutics.
Introduction
In many cancers, platelet interactions with circulating tumor cells and their role in metastasis have been studied extensively.1,2 Murine models of ovarian cancer suggest that platelets undergo extravasation into the primary ovarian tumor and that the platelets promote cancer cell proliferation and increase the resistance of cancer cells to chemotherapy.3-6 However, although the processes governing immune cell trafficking into the tumor microenvironment is better understood,7 how platelets communicate with a vessel under flow, traverse its endothelium, and reach the tumor remain to be elucidated. The mechanisms that regulate platelet transvascular transport in cancer may largely depend on a triad comprising cellular and molecular interactions among the ovarian cancer cells, endothelium, and platelets. Studies relating the vascular consequences of cancer and vice versa suggest that tumors remodel blood vessels, and their released products may compromise vessel barrier integrity,8 making way for extravasation of platelets into the tumor microenvironment.9-11 Therefore, it may be speculated that protecting the endothelial barrier from a tumor eventually arrests interactions with platelets that make cancer cells more potent. To objectively test these hypotheses and dissect the events underlying human platelet extravasation with animal models is not easily possible.12 For example, determining vascular alterations near cancer tissue over time is extremely challenging, and differential tissue and cell effluents cannot be continually collected for downstream genetic and proteomic analyses. Two-dimensional culture plates also do not offer 3-dimensional (3D) organ-level microenvironment and flow.
This study represents the first proof of concept that vascular regulation of platelet extravasation in ovarian cancer can be modeled with organ-on-a-chip (organ chip) technology. An organ chip is a microfluidic cell culture platform with multicellular compartments that mimic the tissue-tissue interface and the mechanical forces that govern biological function.13 This platform offers an in vitro tool for studying how cells and molecules work alone or in combination to influence human disease and progression. In our past work, we have shown that vascular organ chips perform quantitative analysis of organ-level contributions to vascular dysfunction, platelet hyperactivity, and thrombosis.14,15 Importantly, our vessel chip methods revealed mechanisms of vascular thrombosis and the drug-endothelial interactions that are seen in clinical trials.16 In cancer research, organ-on-a-chip technology has spurred investigations in cancer metastasis, intravasation, extravasation, and angiogenesis that are not easily or reliably possible with other models.17-19 Organ chips in cancer hematology research are promising because of to their inherent potential for modeling the tumor microenvironment close to human in vivo conditions. More recently, this method was deployed to reveal pathological interactions between pancreatic cancer cells and endothelial cells.20 The integrative nexus of cancer, vascular biology, and hematology has not been modeled thus far. We have designed a tumor-vessel-blood–integrated organ chip (the OvCa-Chip) that provides insight into the time-dependent endothelial activity that occurs near ovarian cancer tissue and, consequently, its contribution to platelet extravasation from the bloodstream into the tumor microenvironment. With this platform and corresponding histology of tumors isolated from patients with ovarian cancer, we developed a therapeutic strategy that eventually prevented platelet extravasation and direct contact with the tumor.
Methods
OvCa-Chip design and tissue culture
The microchip was designed and fabricated by using a previously described protocol.21 In brief, 2 microfluidic chambers (adjacent parallel tumor channel and blood vessel channel) were formed (top tumor channel: 400 µm wide, 100 µm high; bottom vascular channel: 400 µm wide, 100 µm high; 20-mm long) using soft lithography of polydimethylsiloxane (PDMS) separated by a collagen-fibronectin–coated porous PDMS membrane (∼8-µm pores). The lower channel was lined with endothelium (5 million to 6 million cells/mL) on all sides of the wall and cultured for 24 hours to form a confluent lumen. After the lumen formed, we seeded the tumor chamber with A2780 ovarian cancer cells (1 × 105 cells/cm2) or human ovarian surface epithelial cells, to form the tumor-vascular interface of the OvCa-Chip and Control-Chip, respectively (section 1.1-1.3; supplemental Methods).
Human platelet isolation and perfusion in the OvCa-Chip
Platelets were prepared from centrifugation of human fresh whole blood. Immunofluorescence labeling of platelets was performed with a phycoerythrin-conjugated CD-41 monoclonal antibody. Platelets near the physiological concentration (200 × 103/µL)22 were perfused inside the vascular chamber of the device by gravity. We initially attached a sterilized microtip at the vascular chamber inlet and outlet and perfused 400 µL of the platelet suspension through, creating a pressure head difference within the tips. We controlled the pressure head between vascular and tumor inlet and outlets. The pressure difference across the vascular and tumor chambers was nearly 10 mm, which yielded the desirable average flow rate of 0.45 µL/min and shear stress of ∼1 dyne ⋅ cm−2.23 After every 3 hours when the fluid menisci in both tips were found to be nearly level, we collected the platelets from the outlet and added them back to the inlet to recreate a pressure drop. After perfusion of the platelets inside the vessel, the device was washed repeatedly with a perfusion of 1× phosphate-buffered saline to remove the unbound platelets. Images of platelets inside the device were obtained by fluorescence and laser scanning confocal microscopy, and image analysis was performed with ImageJ software (section 1.4-1.5; supplemental Methods).
Vascular endothelial barrier function
The barrier function was assessed by fluorescein isothiocyanate dextran dye diffusion assay and subsequent confocal imaging of the vascular microchannel. Vessel permeability was quantified by measuring the mean fluorescence intensity (MFI) of the dye that diffused into the collagen-fibronectin layer. The layer of collagen-fibronectin around the vessel wall was selected as the area of interest, and the MFIs of the OvCa-Chip and the Control-Chip were compared and represented as a ratio to determine the relative barrier function. Gaps were measured with confocal images of the endothelial junctions, which were processed by using color thresholding and particle analysis in ImageJ (section 1.6-1.7; supplemental Methods).
Cytokines
The media effluents collected during different hours from the vascular channels of the OvCa-Chip and Control-Chip were analyzed for 27 cytokines, by using the Milliplex Map Human Cytokine/Chemokine Magnetic Bead Panel Kit (CAS-Hcytomag-60 K; Millipore) containing the beads tagged with specific antibodies against the target cytokines. The assays were run in triplicate for each sample, to confirm the concentration of the cytokines identified. Because the sample volume obtained from a single device was not sufficient in some cases, we collected effluents from 8 devices running in parallel and performed the assay as instructed by the manufacturer (section 1.8; supplemental Methods).
Gene expression studies
Expression of endothelial cell genes was measured by quantitative real time-polymerase chain reaction (qRT-PCR). Total cellular RNA was isolated from cells by using a specific kit followed by cDNA synthesis. cDNA (0.5 µg) was used for each qRT-PCR reaction with gene-specific primers, keeping GAPDH as the internal control (section 1.9; supplemental Methods).
Drugs
Atorvastatin was dissolved in 100% ethanol and prepared as a 1-mM stock solution, which was further diluted in EGM-2 medium before perfusion into the vascular lumen to obtain a range of working concentrations (1-100 nM).24 The ethanol concentration was corrected accordingly with a serial dilution and was identical in the drug preparations applied.
Flow cytometry
Vascular endothelial cells isolated from the device were fixed (2% paraformaldehyde), permeabilized (0.1% Triton X100) and then immunostained for VE-cadherin and Src proteins before fluorescence-activated cell sorting. The phosphorylated tyrosine of endothelial cell Src was immunostained with anti-phosphotyrosine antibody. Flow cytometry data acquisition and analyses were performed with an Accuri C6 flow cytometer (BD Biosciences), and data were processed with CellQuest Pro software.
Src phosphorylation assay
Phosphorylation of Src in endothelial whole-cell lysate was measured with Magpix instrumentation (Luminex Corp. Inc., Austin, TX) and a Human Src panel kit (Milliplex 48-650Mag; Merck Millipore). The assays were run in triplicate for each sample, to confirm the Src phosphorylation according to the manufacturer’s protocol, and results were reported as the MFI.
Immunohistochemistry
Immunohistology of healthy or patient-derived ovarian tissue sections were performed with standard fixation and immunostaining methods. Ovarian tumor cell nuclei were stained with hematoxylin, and platelets were stained for CD42b surface proteins. Images were obtained with a microscope, and image analyses were performed to quantify platelet extravasation into the tumors (section 1.10; supplemental Methods).
Statistical analysis
Data are reported as means ± standard error of the mean (SEM) and were analyzed with the tests specified in the figure legends.
Human blood samples were used according to the policies of the US Office of Human Research Protections and approved by the Texas A&M University Institutional Review Board (ID: IRB2016-0762D). Tissue samples were used, and the study was approved by The University of Texas MD Anderson Cancer Center Institutional Review Board.
Results
Establishing the OvCa-Chip
We set out to create the tumor vessel-tissue interface of human ovarian cancer with organ-on-a-chip methodology and created the OvCa-Chip (Figure 1A-C). The anatomy of the OvCa-Chip consisted of 2 overlaid microfluidic chambers separated by matrix-coated porous membrane, as we have shown before.25,26 Each chamber had a width of 200 µm, with a height of 100 µm, and a length of 20 mm (Figure 1D). In this study, we cultured A2780 epithelial ovarian cancer cells in the upper chamber of the OvCa-Chip, because this cell line had been shown to promote platelet extravasation in vivo in a prior study,27 and the lower chamber comprised an endothelial lumen made of human umbilical vein endothelial cells (HUVECs; Figure 1E). At all times, the medium perfusion was maintained at ∼0.5 µL/min inside the vessel, which provided a typical ovarian microvascular shear stress of ∼1 dyne ⋅ cm−2.27 After coculture of these cells for 6 hours, we observed A2780 cancer cells interfacing closely with the HUVECs in the chip (Figure 1F). By 24 hours, the A2780 cells formed a characteristic spheroidal cellular morphology (∼40 μm diameter) in the upper chamber. Immunostaining for cytosolic ZO-1 protein revealed the distribution of the cells with a characteristic 3D spheroidal growth adjacent to the vascular lumen (Figure 1F). In the lower chamber, HUVECs formed confluent monolayers on all sides of the vascular chamber comprising tight cell-cell adhesions, as seen through VE-cadherin expression and characteristic spindle-shaped morphology (25-40 μm diameter; supplemental Figure 1). We also did not observe A2780/HUVEC colocalization or any prominent cancer cell intravasation or transdifferentiation across vessel walls in this assembly. These data showed that we could form a cancer vessel-tissue interface on a chip that is capable of platelet perfusion and analysis of events that may lead to platelet extravasation from the vascular chamber to the adjacent cancer tissue.

Organ-on-a-chip model of ovarian cancer vessel-platelet cross talk. (A) A human cancerous ovary with an injured blood vessel adjacent to tumors, with transmigrating blood cells. This interface is modeled with the OvCa-Chip. (B-C) Cross section of an OvCa-Chip. Two fluidic chambers (red, vessel; blue, cancer cells) in an adjacent superimposed position. (D) Computer-aided design showing the components of OvCa-Chip assembly in detail. The device was fabricated with PDMS soft lithography, with overlaid microchannel compartments bonded to each other with a microporous PDMS membrane between. (E) Microscopy of OvCa-Chip (center) showing human ovarian A2780 tumor cells cultured on the upper microchannel (top) and a human primary-endothelial-cell–formed vessel inside the lower channel (bottom). Bars represent 100 µm. (F) Confocal fluorescence micrograph shows a cross section of the OvCa-Chip, with spheroidal A2780 ovarian cancer cells (red, anti-ZO1) cocultured with primary endothelial cells (green, VE-cadherin) that form continuous monolayers and cover all 4 sides of the lower microchannel, creating a blood-perfused vessel. The tumor-vascular tissue interface distance shown in the cross-sectional 3D view is 10 µm. Platelets are perfused into the vascular lumen of the chip. Bars represent 100 µm.
Organ-on-a-chip model of ovarian cancer vessel-platelet cross talk. (A) A human cancerous ovary with an injured blood vessel adjacent to tumors, with transmigrating blood cells. This interface is modeled with the OvCa-Chip. (B-C) Cross section of an OvCa-Chip. Two fluidic chambers (red, vessel; blue, cancer cells) in an adjacent superimposed position. (D) Computer-aided design showing the components of OvCa-Chip assembly in detail. The device was fabricated with PDMS soft lithography, with overlaid microchannel compartments bonded to each other with a microporous PDMS membrane between. (E) Microscopy of OvCa-Chip (center) showing human ovarian A2780 tumor cells cultured on the upper microchannel (top) and a human primary-endothelial-cell–formed vessel inside the lower channel (bottom). Bars represent 100 µm. (F) Confocal fluorescence micrograph shows a cross section of the OvCa-Chip, with spheroidal A2780 ovarian cancer cells (red, anti-ZO1) cocultured with primary endothelial cells (green, VE-cadherin) that form continuous monolayers and cover all 4 sides of the lower microchannel, creating a blood-perfused vessel. The tumor-vascular tissue interface distance shown in the cross-sectional 3D view is 10 µm. Platelets are perfused into the vascular lumen of the chip. Bars represent 100 µm.
Dynamics of platelet extravasation in an OvCa-Chip
Because platelet extravasation to solid tumors has been observed in murine models,3,27 we set out to determine whether similar pathophysiological consequences of ovarian cancer could be recapitulated with the OvCa-Chip. Therefore, once we had formed the OvCa-Chip with tumor-endothelial coculture over 48 hours, we perfused the vascular lumen of the device with platelets (∼200 × 103/µL) freshly isolated from human blood for the next 5 days (Figure 2A). We leveraged the strength of our system to evaluate physiological activity over time on the device through longitudinal microscopy and quantitated platelet extravasation potential toward cancer cells every day (24 hours). Over the period of 5 days of platelet perfusion, we found consistent expression of P-selectin on the platelets, and they were activated within the microenvironment of the OvCa-Chip (supplemental Figure 2). Three days after platelet perfusion, we observed adhesion of platelets on the surface of cancer-influenced endothelial cells of the OvCa-Chip, but no platelets were observed in the Control-Chip, in which cancer cells were absent (Figure 2B-E). We also saw platelets colonized into a tumor, thus confirming that, similar to the in vivo situation, platelets extravasated from the vascular lumen into the tumor compartment within our OvCa-Chips (Figure 2F; supplemental Video 1). Quantification of adherent platelets revealed that, from 72 hours onward, there was rapid acceleration in platelet adhesion (Figure 2G). Correspondingly, the platelets attached to the endothelium eventually crossed the barriers and migrated into the tumor compartment during the same time frame (Figure 2H). When the endothelium was completely removed, platelet extravasation was at its maximum when the tumor cells were present, but nearly absent when replaced with benign cells (Figure 2I), suggesting that platelet extravasation is controlled by gain or loss of endothelium. These data showed that our OvCa-Chip could model platelet extravasation dynamics and provide a tool to evaluate how ovarian cancer cells influence the vascular luminal microenvironment and trigger platelet extravasation and subsequent migration toward the lumina over time. Because it is widely known that immune cell trafficking occurs in cancer and vascular barrier function is compromised, these observations in organ chips prompted us to hypothesize that platelet extravasation is triggered by ovarian cancer cells through systematic and dynamic disruption of the neighboring endothelium. Consequently, we performed the following experiments to explore the cancer vessel signaling that eventually results in platelet extravasation in OvCa-Chips.

Platelet extravasation in the OvCa-Chip. (A) Time line describing the events and end points when endothelial cells, tumor cells, and platelets are introduced into the chip, followed by analysis of platelet extravasation. The tumor-endothelial coculture was established in the first 48 hours. Platelet perfusion was initiated at 48 hours, and the vascular alteration and subsequent platelet extravasation physiology was observed for up to 168 hours (7 days). Confocal micrographs showing 3D cross-sectional views of the OvCa-Chip consisting of cancer cells (blue, nuclei), vascular endothelial lumen (green, VE cadherin), and platelets (red, anti CD41) (B) and the Control-Chip consisting of control cells (blue), vascular endothelial lumen (green), and platelets (red) (C), every 24 hours after the platelets were introduced into the device. Bars represent 100 µm. (D) Confocal micrographs showing platelets (red) adhering to the vascular endothelium (Endo, green) and corresponding extravasation of platelets into the cancer cells (Cancer cells, blue) crossing the dysfunctional vascular barrier, at increasing time points when cancer cells were cocultured within OvCa-Chip. Bars represent 25 µm. (E) No prominent platelet adherence to the endothelial cells (Endo, green) and extravasation into control cells (Control cells, blue) was found in the Control-Chip. (F) After 72 hours, time-lapse images (brightfield and fluorescence overlaid; 10 minutes apart) taken within the tumor chamber showing platelets (red) extravasating from the luminal side (arrow) through the pore and entering into the cancer tissue compartment. Bar represents 25 µm. The quantitation of platelet adherence to the endothelium (G); extravasated platelet concentration observed within the tumor chamber (H) (●, OvCa-Chip; ▪, Control-Chip); and extravasated platelet concentration because of gain or loss of function of endothelial cells (I). Shaded area in panels G and H represents the 48-hour period before the introduction of platelets into the OvCa-Chip, when tumor-vessel coculture was established. Error bars indicate the mean ± SEM; n = 3 individual experiments. Two-tailed paired Student t test. *P < .05; **P < .01. ns, nonsignificant.
Platelet extravasation in the OvCa-Chip. (A) Time line describing the events and end points when endothelial cells, tumor cells, and platelets are introduced into the chip, followed by analysis of platelet extravasation. The tumor-endothelial coculture was established in the first 48 hours. Platelet perfusion was initiated at 48 hours, and the vascular alteration and subsequent platelet extravasation physiology was observed for up to 168 hours (7 days). Confocal micrographs showing 3D cross-sectional views of the OvCa-Chip consisting of cancer cells (blue, nuclei), vascular endothelial lumen (green, VE cadherin), and platelets (red, anti CD41) (B) and the Control-Chip consisting of control cells (blue), vascular endothelial lumen (green), and platelets (red) (C), every 24 hours after the platelets were introduced into the device. Bars represent 100 µm. (D) Confocal micrographs showing platelets (red) adhering to the vascular endothelium (Endo, green) and corresponding extravasation of platelets into the cancer cells (Cancer cells, blue) crossing the dysfunctional vascular barrier, at increasing time points when cancer cells were cocultured within OvCa-Chip. Bars represent 25 µm. (E) No prominent platelet adherence to the endothelial cells (Endo, green) and extravasation into control cells (Control cells, blue) was found in the Control-Chip. (F) After 72 hours, time-lapse images (brightfield and fluorescence overlaid; 10 minutes apart) taken within the tumor chamber showing platelets (red) extravasating from the luminal side (arrow) through the pore and entering into the cancer tissue compartment. Bar represents 25 µm. The quantitation of platelet adherence to the endothelium (G); extravasated platelet concentration observed within the tumor chamber (H) (●, OvCa-Chip; ▪, Control-Chip); and extravasated platelet concentration because of gain or loss of function of endothelial cells (I). Shaded area in panels G and H represents the 48-hour period before the introduction of platelets into the OvCa-Chip, when tumor-vessel coculture was established. Error bars indicate the mean ± SEM; n = 3 individual experiments. Two-tailed paired Student t test. *P < .05; **P < .01. ns, nonsignificant.
Degradation of endothelium in OvCa-Chips
Previous studies have demonstrated that cancer cells may induce structural disturbances in a nearby blood vessel by affecting its endothelial cell-cell adherence junctions.8 We postulated that platelet extravasation from the vessel into the ovarian tumors would be a direct consequence of endothelial degradation. Therefore, we sought to investigate the impact of ovarian cancer cells on the vascular endothelial lumen when cocultured within the OvCa-Chip for 5 days (120 hours), in the absence of platelets. In the first 48 hours, we formed the OvCa-Chip with intact vascular lumen cocultured with tumor cells as described earlier (Figure 1). We were able to see that the vascular lumen was intact for another 48 hours of coculture with A2780s cells except a few minor gaps. At 72 hours, we saw a decline in VE-cadherin expression (Figure 3A-D; supplemental Video 2) and an increase in gaps (Figure 3E; supplemental Figure 3) and barrier disruption (Figure 3F; supplemental Figure 4), whereas the Control-Chip showed no such effect. The degradation continued to increase for 120 hours, at which time, we saw significant loss in the barrier function of the endothelial lumen and nearly complete destruction of the OvCa-Chip, possibly because of overgrowth and the inability of the medium to supply nutrients in the microfluidic chamber. Importantly, this time line at which barrier degradation occurs in the OvCa-Chip also correlated with the observed platelet extravasation (Figure 2). To increase confidence in our results, we conducted similar experiments with an OvCa-Chip (version 2) consisting of coculture of another human primary tumor cell type, OVCAR3, and a lumen composed of human ovarian microvascular endothelial cells (HOMECs). As before, we observed vascular adhesion junction degradation and increased gap formation with time, in this second version of the OvCa-Chip (supplemental Figure 5A-B). These data confirmed that our platform models the tumor-vascular interaction dynamics over a period of 5 days and the products released by ovarian cancer cells cause vascular disruption in a time-dependent manner.
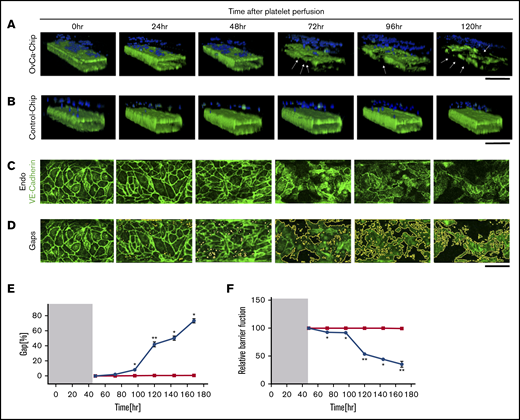
Vascular degradation of the OvCa-Chip. Confocal micrographs showing 3D cross-sectional views of the OvCa-Chip, consisting of cancer cells (blue, nuclei) and vascular endothelial lumen (green, VE-cadherin) (A), and the Control-Chip, consisting of benign control cells (blue, nuclei) and vascular endothelial lumen (green, VE-cadherin) (B), imaged every 24 hours after cancer cells were introduced into the device. Arrows: degradation of endothelium and the gaps formed as a result. Bars represent 200 µm. High-resolution confocal micrographs of the vascular endothelial monolayer showing distinct loss of VE-cadherin (green) (C) and subsequent formation of gaps over time (identified in image processing) (D). Bars represent 25 µm. The distinct increase in vascular endothelial gaps (E), and the loss over time of vascular barrier function (F) in OvCa-Chips (●) relative to Control-Chips (▪). Shaded areas in panels E and F represent the 48-hour period when tumor-vessel coculture was established. Error bars indicate the mean ± SEM; n = 3 individual experiments. Two-tailed paired Student t test. *P < .05; **P < .01.
Vascular degradation of the OvCa-Chip. Confocal micrographs showing 3D cross-sectional views of the OvCa-Chip, consisting of cancer cells (blue, nuclei) and vascular endothelial lumen (green, VE-cadherin) (A), and the Control-Chip, consisting of benign control cells (blue, nuclei) and vascular endothelial lumen (green, VE-cadherin) (B), imaged every 24 hours after cancer cells were introduced into the device. Arrows: degradation of endothelium and the gaps formed as a result. Bars represent 200 µm. High-resolution confocal micrographs of the vascular endothelial monolayer showing distinct loss of VE-cadherin (green) (C) and subsequent formation of gaps over time (identified in image processing) (D). Bars represent 25 µm. The distinct increase in vascular endothelial gaps (E), and the loss over time of vascular barrier function (F) in OvCa-Chips (●) relative to Control-Chips (▪). Shaded areas in panels E and F represent the 48-hour period when tumor-vessel coculture was established. Error bars indicate the mean ± SEM; n = 3 individual experiments. Two-tailed paired Student t test. *P < .05; **P < .01.
Cytokine expression in OvCa-Chip
It has been shown that cancer cells secrete inflammatory cytokines that may disrupt vascular integrity.8 Further, the cytokines may influence the endothelium to exert a cumulative effect that results in vascular inflammation and structural disturbances.28 Therefore, in ovarian cancer, it is highly likely that the cumulative effect of cytokines released from cancer cells and endothelium creates endothelial discontinuity and a proinflammatory microenvironment favorable to platelet extravasation. Because it is difficult to analyze several cytokines together at various time points in vivo and because the organ chips emitted a nearly continuous collection of effluents and multiplexed cytokine readouts, we set out to retrieve the effluents from the OvCa-Chips at several time points over 5 days (120 hours) and to analyze the concentrations of 27 major human cytokines (Figure 4A; supplemental Figure 6; supplemental Table 1). We found that 4 cytokines (IL-8, MCP-1, IL-6, and TNF-α) were present in high concentrations in the effluents available from the OvCa-Chip, compared with those from Control-Chip, and interestingly, their concentrations increased significantly during 48 to 96 hours of cancer vessel coculture, which corresponds to the disruption of vascular integrity and platelet extravasation that was observed in the same time line. These peaks of cytokines were followed by an eventual decrease at the 120-hour measurement point, correlating with the rapid apoptosis of cancer and endothelial cells observed inside the device. We also analyzed the cytokine profile in the OvCa-Chip (version 2) consisting of a OVCAR3-HOMEC coculture and found a major increase in the cytokines (MCP-1, IL6, IL-8, and TNA-α), as observed previously, that can influence the endothelium, along with some others (supplemental Figure 5C). Therefore, our data demonstrate that organ chips can indeed model the proinflammatory vascular microenvironment near ovarian tumors and reveal time-dependent secretion of factors that may regulate ovarian cancer progression.

Cytokines and gene expression analysis. (A) Concentrations of cytokines released within the OvCa-Chip relative to the Control-Chip (MCP-1, IL-8, TNF-α, and IL-6), which showed a significant increase in time (n = 3 individual experiments). (B) Relative expression of different endothelial genes in OvCa-Chips vs Control-Chips. The percentage expression of respective genes (relative to GAPDH) were normalized with respect to the percentage expression measured in Control-Chips. Error bars indicate the mean ± SEM; n = 3 individual experiments. Two-tailed paired Student t test; *P < .05.
Cytokines and gene expression analysis. (A) Concentrations of cytokines released within the OvCa-Chip relative to the Control-Chip (MCP-1, IL-8, TNF-α, and IL-6), which showed a significant increase in time (n = 3 individual experiments). (B) Relative expression of different endothelial genes in OvCa-Chips vs Control-Chips. The percentage expression of respective genes (relative to GAPDH) were normalized with respect to the percentage expression measured in Control-Chips. Error bars indicate the mean ± SEM; n = 3 individual experiments. Two-tailed paired Student t test; *P < .05.
Endothelial signaling pathways in the OvCa-Chip
In several disease models, secretion of inflammatory cytokines has been suggested to activate vascular endothelial cell Src/ERK/FAK signaling pathways to disrupt cell-cell adhesion junctions (VE-cadherin–β-catenin cytoskeletal complex) and increase the permeability of blood vessels.8,28-34 However, in ovarian cancer, these pathways have been relatively underexplored in the context of tumor-platelet interactions. Therefore, we sought to test the hypothesis that in the OvCa-Chip, cancer cells activate the endothelial Src/ERK/FAK kinase signaling cascade, which downregulates the expression of the VE-cadherin/β-catenin complex and upregulates Akt, thereby inducing vascular endothelial adhesion gap formation. To test this hypothesis, we first collected the endothelial cells from the OvCa-Chips (or Control-Chips) after 72 hours. Then, we set out to measure the gene expression profile of the Src/ERK/FAK cascade inside the endothelial cells through quantitative real-time PCR (Figure 4B; supplemental Table 2). We found a significant increase in the expression of the Src and FAK genes and a modest increase in ERK (possibly, because ERK is relatively conserved within HUVECs) in the OvCa-Chip, which indicated that they regulated vascular barrier disruption. Correspondingly, we found a significant downregulation in the expression of the VE-cadherin and β-catenin genes, as well as VE-PTP (VE-cadherin/β-catenin assembly), the master regulators of vascular barrier function.9,35,36 We also found an increased expression of endothelial Akt in the presence of ovarian cancer cells in our model, which has also been reported to be activated in some metastatic cancers and contributes to cancer growth and endothelial survival.37,38 To further establish the possibility of endothelial activation in ovarian cancer that may regulate platelet extravasation, we also quantitated the expressions of vascular endothelial Tie-2, Pyk-1, and Rac-1 genes, which are known to directly influence activation and transvascular migration of blood leukocytes and neutrophils.32 We found that their expression was significantly increased in the OvCa-Chip relative to that in the Control-Chip, indicating their involvement in the endothelial activation needed for platelet extravasation. These results strongly confirm that the vascular barrier degradation that we observed within the OvCa-Chip is supported by the genetic activation of vascular endothelial signaling pathways (Src/ERK/FAK), attenuation of vascular integrity (VE-cadherin–β-catenin expression), and overexpression of prostimulatory signals for communication with blood cells (Pyk-1/Tie-2/Rac-1). Further, these data demonstrate our system’s ability to analyze several corresponding genetic signatures in a complex disease model consisting of multitissue interactions, which may be difficult to perform with in vivo models.
Atorvastatin therapy prevents platelet extravasation and its consequences in the OvCa-Chip
Statins, such as atorvastatin, have been extensively shown to preserve the integrity of endothelial adherence junctions.28 In several cancers, prescription of statins has improved clinical outcomes in many patients.34,39 Exactly how statins help patients with ovarian cancer is still not fully known and, specifically, their effect on platelet extravasation into tumors has not been reported. Because our OvCa-Chip models tumor-led vascular degradation and corresponding platelet extravasation, we examined whether atorvastatin arrests platelet extravasation by reducing vascular degradation in the OvCa-Chip (Figure 5A). To test this hypothesis, we kept the vascular compartment of the OvCa-Chip in perfused medium but added atorvastatin (10 nM) after 2 days (48 hours) of coculture. Then, we correspondingly measured the status of cell adherence junctions of endothelial cells, luminal cytokine profiles, and the expression of endothelial genes that we had already found to regulate the vascular assembly. We found that in the absence of atorvastatin treatment within the OvCa-Chip, adhesion junctions (VE-cadherin) were disturbed, and endothelial gaps formed on the endothelial lumen, as expected. In contrast, the atorvastatin-treated OvCa-Chip exhibited significantly improved junction integrity and reduced gaps (Figure 5B-C; supplemental Video 3). Next, when we quantified the vascular effluents available from the atorvastatin-treated OvCa-Chip, we found a distinct reduction in the cytokines MCP-1, IL-8, TNF-α, and IL-6, which were found to be crucial for endothelial degradation of this tumor-vessel interface (Figure 5D). Gene expression analysis of vascular endothelium obtained from the chip revealed a time-dependent but distinct reduction in the expression of Src, ERK, and FAK; an increase in VE-cadherin, β-catenin, and VE-PTP; and decreases in Tie-2, Pyk-1, and Rac-1, nearly similar to observations in the Control-Chip (Figure 5E). Some prior studies have shown that statins activate Akt to promote angiogenesis and regeneration.40,41 Corresponding to these studies, our model showed that Akt expression in a drug OvCa-Chip was further increased, and endothelial Akt was more highly activated relative to that in the OvCa-Chip. Altogether, these data suggest that upon treating the endothelium with atorvastatin, the microvascular environment near cancer cells can be prevented from becoming significantly leaky and proinflammatory, and promote their survival. In addition, atorvastatin applied to the Control-Chip did not affect the morphology of the endothelial lumen at a dose of 10 nM, but, as expected, when a higher atorvastatin dose (100 nM) was applied to the Control-Chip, we found a morphological disturbance in the endothelial cells within 6 hours and a rapid adherence of platelets, revealing an opposite and toxic effect at a 10-fold higher dose (supplemental Figure 7A-B). Also, a low concentration (1 nM) of the drug applied to the OvCa-Chip remained ineffective in causing restoration of vascular adherence (supplemental Figure 7C). Further, we specifically investigated the proteomic status of Src kinase, to support the gene expression analysis. To show whether Src kinase is upregulated when the barrier is compromised, we added IL-8 (5 ng/mL) inside the Control-Chip, an inflammatory cytokine that induces disturbance in vascular barrier integrity. After the typical 48-hour time point, we observed a rapid reduction in VE-cadherin expression that resulted in the loss of the endothelial adhesion junction (supplemental Figure 8). In this analysis, we saw a corresponding upregulation of endothelial Src kinase and its rapid phosphorylation. Further, we found that, in correlation with loss of VE-cadherin, there was high expression of Src in the OvCa-Chip, relative to that in the Control-Chip (Figure 6A-C). Interestingly, this expression was downregulated with atorvastatin. In addition, we detected rapid phosphorylation (activation) of Src in tumor-influenced endothelial cells, and this activity was correspondingly suppressed with atorvastatin therapy, when characterized by immunofluorescence, flow cytometry, and protein kinase phosphorylation assays. These data support the activation of Src kinase pathway correlating with our previously obtained gene expression data. Therefore, OvCa-Chip recapitulated atorvastatin’s effect on the endothelium. Finally, to demonstrate a broader capability and as a measure of comparison, in addition to atorvastatin, we also sought to determine the therapeutic potential of simvastatin. When we treated the OvCa-Chip with 10 nM simvastatin, we again observed partial restoration of the adherence junction (supplemental Figure 9). The results showed that our organ-on-a-chip predicted the extent of therapeutic effect of atorvastatin on the vascular endothelium interfaced with ovarian cancer cells from tissue, to cell, to gene level and provided some critical insight into the multifactorial drug-tissue interactions. Next, when we perfused an OvCa-Chip that had added atorvastatin with platelets, we observed a reduced number of platelets adhering to the endothelium and their extravasation into the cancer cell compartment, compared with those in platelet-infused Control-Chips (Figure 7A-D). Therefore, our data predict that statins potentially serve as drugs that prevent cancer growth, partially by arresting their direct contact with platelets.

Evaluation of atorvastatin therapy in the OvCa-Chip. (A) Hypothesized signaling and critical molecular events that lead to vascular degradation and platelet extravasation predicted by the OvCa-Chip. It was expected that these pathways and functional consequences would be arrested by atorvastatin treatment through changes in expression of proinflammatory cytokines and genes, such as Src/FAK/ERK, Tie-2, Pyk-1, Rac-1, Akt, and VE-cadherin/β-catenin/VE-PTP. (B) Confocal micrographs showing vascular endothelial junctions (VE-cadherin) in the Control-Chip (top), OvCa-Chip (center), and OvCa-Chip treated with atorvastatin (bottom). Bar represents 200 µm. Quantification of endothelial gaps (C) and expression of major inflammatory cytokines (D) (inset: IL-6 concentration at a reduced scale) released in OvCa-Chip relative to the Control-Chip. (E) Heat map generated from qRT-PCR data showing normalized time-dependent gene expression analysis within Control-Chip, OvCa-Chip, and OvCa-Chip treated with atorvastatin. One-way ANOVA, followed by Dunnett’s multiple-comparisons test. Error bars are the mean ± SEM; n = 3 individual experiments. *P < .05; **P < .01.
Evaluation of atorvastatin therapy in the OvCa-Chip. (A) Hypothesized signaling and critical molecular events that lead to vascular degradation and platelet extravasation predicted by the OvCa-Chip. It was expected that these pathways and functional consequences would be arrested by atorvastatin treatment through changes in expression of proinflammatory cytokines and genes, such as Src/FAK/ERK, Tie-2, Pyk-1, Rac-1, Akt, and VE-cadherin/β-catenin/VE-PTP. (B) Confocal micrographs showing vascular endothelial junctions (VE-cadherin) in the Control-Chip (top), OvCa-Chip (center), and OvCa-Chip treated with atorvastatin (bottom). Bar represents 200 µm. Quantification of endothelial gaps (C) and expression of major inflammatory cytokines (D) (inset: IL-6 concentration at a reduced scale) released in OvCa-Chip relative to the Control-Chip. (E) Heat map generated from qRT-PCR data showing normalized time-dependent gene expression analysis within Control-Chip, OvCa-Chip, and OvCa-Chip treated with atorvastatin. One-way ANOVA, followed by Dunnett’s multiple-comparisons test. Error bars are the mean ± SEM; n = 3 individual experiments. *P < .05; **P < .01.

Analysis of endothelial Src kinase in the OvCa-Chip. (A) Confocal micrographs showing the status of endothelial cell adhesion junction VE-cadherin (green), expression of Src kinase (red), and its activation (phosphorylated, yellow) in the Control-Chip, OvCa-Chip, and Drug-OvCa-Chip. Bar represents 25 µm. Nuclei are stained with 4′,6-diamidino-2-phenylindole (blue). Fluorescence-activated cell sorting analysis of immunostained endothelial cells showing VE-cadherin, Src expression, and Src phosphorylation (B), and phosphorylation analysis of endothelial Src kinase by Luminex assay (C), obtained from the Control-Chip, OvCa-Chip, and Drug-OvCa-Chip. Error bars are the mean ± SEM. One-way ANOVA, followed by Dunnett’s multiple-comparisons test; n = 3 individual experiments. *P < .05.
Analysis of endothelial Src kinase in the OvCa-Chip. (A) Confocal micrographs showing the status of endothelial cell adhesion junction VE-cadherin (green), expression of Src kinase (red), and its activation (phosphorylated, yellow) in the Control-Chip, OvCa-Chip, and Drug-OvCa-Chip. Bar represents 25 µm. Nuclei are stained with 4′,6-diamidino-2-phenylindole (blue). Fluorescence-activated cell sorting analysis of immunostained endothelial cells showing VE-cadherin, Src expression, and Src phosphorylation (B), and phosphorylation analysis of endothelial Src kinase by Luminex assay (C), obtained from the Control-Chip, OvCa-Chip, and Drug-OvCa-Chip. Error bars are the mean ± SEM. One-way ANOVA, followed by Dunnett’s multiple-comparisons test; n = 3 individual experiments. *P < .05.

Platelet extravasation is arrested in the OvCa-Chip treated with atorvastatin. Confocal micrographs showing platelet (red) adhering to the vascular endothelium (Endo, green, VE-cadherin) (A), and corresponding extravasation of platelets into the cancer cells (cancer cells, blue, nuclei) (B) crossing the dysfunctional vascular barrier at increasing time points within OvCa-Chip (top) or OvCa-Chip treated with atorvastatin (Drug-OvCa-Chip; bottom). Bars represents 50 µm. Graph showing the quantitation of platelet adherence to the endothelium (C), and extravasated platelet concentration observed within the tumor chamber (D) (▪, OvCa-Chip; ▲, Drug-OvCa-Chip; and ●, Control-Chip). The shaded areas represent the 48-hour period before the introduction of platelets within OvCa-Chip when tumor-vessel coculture was established. P-values calculated vs Control-Chip; n = 3 individual experiments. (E) A representative immunohistochemical analysis of tumor tissue specimens obtained from healthy women and women with ovarian cancer showing platelets extravasated (CD42b+, brown, denoted by arrows) into the tumor stroma, in patients without statin drug treatment vs those with atorvastatin treatment. The bar represents 50 µm. Insets: original magnification ×40, of select regions. (F) Quantification of platelets observed with histology. Error bars are means ± SEM; n = 5 individual patient samples per group. One-way ANOVA, followed by Dunnett’s multiple-comparisons test. *P < .05; **P < .01.
Platelet extravasation is arrested in the OvCa-Chip treated with atorvastatin. Confocal micrographs showing platelet (red) adhering to the vascular endothelium (Endo, green, VE-cadherin) (A), and corresponding extravasation of platelets into the cancer cells (cancer cells, blue, nuclei) (B) crossing the dysfunctional vascular barrier at increasing time points within OvCa-Chip (top) or OvCa-Chip treated with atorvastatin (Drug-OvCa-Chip; bottom). Bars represents 50 µm. Graph showing the quantitation of platelet adherence to the endothelium (C), and extravasated platelet concentration observed within the tumor chamber (D) (▪, OvCa-Chip; ▲, Drug-OvCa-Chip; and ●, Control-Chip). The shaded areas represent the 48-hour period before the introduction of platelets within OvCa-Chip when tumor-vessel coculture was established. P-values calculated vs Control-Chip; n = 3 individual experiments. (E) A representative immunohistochemical analysis of tumor tissue specimens obtained from healthy women and women with ovarian cancer showing platelets extravasated (CD42b+, brown, denoted by arrows) into the tumor stroma, in patients without statin drug treatment vs those with atorvastatin treatment. The bar represents 50 µm. Insets: original magnification ×40, of select regions. (F) Quantification of platelets observed with histology. Error bars are means ± SEM; n = 5 individual patient samples per group. One-way ANOVA, followed by Dunnett’s multiple-comparisons test. *P < .05; **P < .01.
Validation of the OvCa-Chip with human ovarian cancer biopsy samples
Based on the preclinical observations described herein, we next asked whether similar observations would be made in human ovarian cancer samples. We obtained high-grade serous ovarian cancer samples from patients who were or were not treated with atorvastatin (5 samples per group). When compared with normal untreated counterparts, tumor samples from those taking atorvastatin revealed a considerably lower extent (∼62%) of platelet extravasation (Figure 7E-F; supplemental Figure 10). Extravasated platelets were observed to populate the extravascular space and stromal region surrounding tumor cells, mainly in tumor specimens from patients who were not taking atorvastatin. This reduction in extravasated platelet numbers is similar with our on-chip findings and establish the present OvCa-Chip’s capability of modeling the drug’s effects on ovarian cancer vascular pathophysiology.
Discussion
These studies demonstrate a new in vitro preclinical research strategy in which the human OvCa-Chip permits visualization and quantitative temporal analysis of organ-level interactions between cancer cells, endothelial cells, and blood cells. This organ-chip technology allows us to dissect complex intercellular signaling that ultimately leads to the extravasation of platelets into the tumor microenvironment. The time-dependent pathophysiology that we predict with this model cannot be expected to be performed at this resolution in animal models. A salient feature of the OvCa-Chip was that it provided tissue products and effluents that we could subsequently examine. We determined the inflammatory cytokines that played a critical role in the regulation of vascular junction integrity and permeability in this model. Because cytokines also activate platelets, our OvCa-Chip results suggest that the upregulated cytokines (IL-8, MCP-1, IL-6 and TNF-α) also positively contribute to platelet extravasation and that inhibition of these factors may therefore arrest this activity. Then, we also determined the temporal pattern of gene expression in cancer cell-activated endothelium, which maintains the vascular junctions (Src/ERK/FAK/Akt), barrier function (VE-cadherin/β-catenin/VE-PTP), and the activation status (Tie-2/Pyk-1/Rac-1) of vessels crucial for platelet extravasation. Significantly, we applied the model to assess dose-dependent effect of atorvastatin in ovarian cancer. Our data showed that the OvCa-Chip may serve to identify mechanisms of cancer cell-endothelial cross talk. For example, our observation of successive activation of endothelial Akt in OvCa-Chip and drug OvCa-Chip indicates that the model can be further deployed to determine the exact mechanism of atorvastatin-mediated upregulation of Akt, which remains largely unknown.42 Further, our finding from the organ chip and concurrent histology of biopsy samples validates the model and provides us with one of the functional mechanisms by which atorvastatin potentially benefits patients with ovarian cancer. Based on our results, we also predict that atorvastatin can serve as a drug that prevents cancer growth, partially by arresting the direct contact of cancer cells with platelets. Our own results shed some light on the mechanism of statins on the endothelial barrier. We showed that Src kinase activation and phosphorylation are specifically involved in maintenance of the endothelial barrier and that statins conserve the endothelial barrier through the Src pathway. Several prior studies of statins have shown that they protect endothelial function through both cholesterol-dependent and -independent functions. In the context of this work, one of the ways that statins improve endothelial barrier function independently is by increasing the bioavailability of nitric oxide (NO), through upregulating the expression of eNOS. Prior work has shown that eNOS-derived NO tightly regulates endothelial barrier function through VE-cadherin.43 Also, a prior study showed that TNF-α incubation of endothelial cells results in a reduction in eNOS protein expression, but statins prevent it and therefore improve endothelial barrier function.44 Similarly, in an inflamed endothelium, a decrease in ρ GTPase response takes place during statin treatment. This relieves the inflammation-induced suppression of eNOS mRNA synthesis and therefore increases the production and bioavailability of endothelium-derived NO.45,46 Therefore, the ρ/ROCK pathways may be a downstream contributor to the pleiotropic effects of statin and may be an important mechanism that also plays a role in inhibiting platelet extravasation in ovarian cancer. Although in this study we focused on platelet extravasation, it is possible that atorvastatin affects the traffic of immune cells to the tumor and alters its immune profile. Also, even though statins were shown to partially protect the barrier and arrest platelet extravasation, they did not fully restore the endothelium. Because platelet activation was observed, it is possible that other therapeutics (for example, antiplatelet drugs) are needed in combination with statins. Therefore, the full potential of the current model has not yet been explored and, like other models, it may not include all the cells of the ovarian tumor microenvironment. In this study, our efforts were directed toward balancing the complexity in the design, including multicellular architecture, critical human tissue-tissue interfaces, and hemodynamics, with simplicity, for example, by not including specific incorporation and analysis of the extracellular matrix environment, even though our cancer vessel interface is occupied by extracellular matrix that may be continuously remodeled by cultured cells. We did not include pericytes or stromal cells that could exert influence on platelet extravasation. We also did not perfuse whole blood in the vascular chamber, because in this study, we maintained flow for 5 days, and it is very difficult, practically, to maintain blood fluidity in vitro for this prolonged period. Nonetheless, by using platelets, we specifically evaluated platelet extravasation independent of the effect of other blood cells. By having the same decrease in pressure in both the tumor and the vessel chambers, we kept the transvascular interstitial pressure nearly equal. There is significant evidence, however, that tumors elevate the interstitial pressure, thus creating an adverse gradient for any transvascular flow to occur in vivo.47 Platelet extravasation under elevated interstitial pressure was beyond the scope of our work, but it would be an interesting investigation that could be systematically conducted with our platform. We expect that this technology will be an advance toward personalized cancer medicine by integrating induced pluripotent stem cells and other patient-derived cell sources, so that we can more faithfully recapitulate more specific responses to therapy.
In summary, our human OvCa-Chip model may offer a platform for testing new hypotheses, acquiring functional readouts, and performing preclinical tests of drugs before embarking on large animal studies and clinical trials of cancer treatments.
The original data are available by e-mail request to the corresponding author, Abhishek Jain (a.jain@tamu.edu).
Acknowledgments
The authors thank J. A. Culpepper (Texas A&M University) for managing the phlebotomy of healthy individuals, S. Vitha (Texas A&M Microscopy and Imaging Center) for assisting with the confocal imaging, P. Biswas for assisting in the MAGPIX experiments, and J. Bui for fabricating the organ chips.
This work was supported by National Institutes of Health (NIH), National Institute of Biomedical Imaging and Bioengineering (NIBIB) (grant R21EB025945); Texas A&M Engineering Experimentation Station (TEES) and Texas A&M University President’s Excellence in Research X-Grant (A.J.). Portions of the work were also supported by the NIH, National Cancer Institute (grants P30 CA016672, P50 CA217685, CA177909, and R35 CA209904); the Anderson Cancer Center Ovarian Cancer Moon Shot Program; the Blanton-Davis Ovarian Cancer Research Program; American Cancer Society Research Professor Award; and the Frank McGraw Memorial Chair in Cancer Research (all A.K.S.); and a training fellowship from the Gulf Coast Consortia and the Computational Cancer Biology Training Program (CPRIT grant RP170593) (K.F.H.).
Authorship
Contribution: B.S. and A.J. designed and performed the experiments, prepared the figures, and wrote the manuscript with support from all other authors; T.M prepared and processed the video files; K.F.H. and W.H. acquired and analyzed patient data to identify and obtain patient biopsy samples for histology, under the direction of A.K.S.; and A.K.S. and V.A.-K. provided ovarian tumor cells, histological slides of tumor tissue and supported in data analysis.
Conflict-of-interest disclosure: A.K.S. served as a consultant for Merck and Kiyatec, is a shareholder in BioPat, and has received research funding from M-Trap. The remaining authors declare no competing financial interests.
Correspondence: Abhishek Jain, Department of Biomedical Engineering, Texas A&M College of Engineering, 101 Bizzell St, College Station, TX 77843; e-mail: a.jain@tamu.edu.

