

Key Points
This article defines elements of adult sickle cell centers to facilitate care and alleviate health disparities in this patient group.
Models of SCD care were developed and disseminated during a workshop to train health care professionals to establish SCD clinical centers.

Abstract
Sickle cell disease (SCD) is the most common inherited blood disorder in the United States. It is a medically and socially complex, multisystem illness that affects individuals throughout the lifespan. Given improvements in care, most children with SCD survive into adulthood. However, access to adult sickle cell care is poor in many parts of the United States, resulting in increased acute care utilization, disjointed care delivery, and early mortality for patients. A dearth of nonmalignant hematology providers, the lack of a national SCD registry, and the absence of a centralized infrastructure to facilitate comparative quality assessment compounds these issues. As part of a workshop designed to train health care professionals in the skills necessary to establish clinical centers focused on the management of adults living with SCD, we defined an SCD center, elucidated required elements of a comprehensive adult SCD center, and discussed different models of care. There are also important economic impacts of these centers at an institutional and health system level. As more clinicians are trained in providing adult-focused SCD care, center designation will enhance the ability to undertake quality improvement and compare outcomes between SCD centers. Activities will include an assessment of the clinical effectiveness of expanded access to care, the implementation of SCD guidelines, and the efficacy of newly approved targeted medications. Details of this effort are provided.
Introduction
Sickle cell disease (SCD) is the most common inherited hemoglobinopathy in the United States, affecting ∼100 000 individuals. Improvements in childhood mortality attributed to universal newborn screening, antipneumococcal vaccination, antibiotic prophylaxis, and disease-specific treatments (eg, blood transfusion and hydroxyurea) have allowed ∼96% of children with SCD to reach adulthood.1 Approximately 60% of the SCD population in the United States is now above the age of 18 years.2
Although most individuals with SCD living in resource-rich countries live into their fifth decade, the burden of illness remains high.3 Excess morbidity and mortality is tied to higher acute care utilization, hospitalization, and readmission rates,1,4,5 especially among patients 18 to 30 years of age.6 A recent modeling study showed a lower projected life expectancy (54 vs 76 years) and quality-adjusted life expectancy (33 vs 67 years) for individuals with SCD relative to those without SCD.7 Data from California are even more concerning: recent estimates put the mean age at death at 43 years for women and 41 years for men with SCD.8
Adults living with SCD are often medically and socially complex and experience significant health disparities.9 These individuals face multiple hurdles to attaining high-quality and continuous care across the lifespan. Upon entering adulthood, patients often lose their primary medical home and health insurance. Historically, SCD was considered a pediatric disease and many adult-focused providers have not received training in its care. Adult patients suffer from a shortage of specialized, knowledgeable providers and difficulties in care coordination between primary and subspecialty services.10,11 In contrast, hemophilia and cystic fibrosis, which each have a population incidence of less than one-half that of SCD in the United States, benefit from >130 comprehensive treatment centers nationwide.9 Furthermore, both conditions have national registries and funding for clinical centers to support their populations whereas SCD does not. For some patients with SCD, especially in low-income and rural communities, the lack of specialized providers and comprehensive care is severe.12,13
The need for comprehensive SCD care has long been recognized, as reflected by the passage of the National Sickle Cell Disease Control Act in 1972. This fueled a National Heart, Lung, and Blood Institute (NHLBI) initiative to create SCD centers with the triple directive of simultaneously embarking upon research, comprehensive care, and engagement with community-based organizations (CBOs) primarily focused on children. This initiative was highly effective at reducing pediatric mortality through newborn screening and penicillin prophylaxis. Unfortunately, funding was not sustained and the centers closed. Although much of the pediatric care was absorbed by academic pediatric hematology-oncology centers, the same is not true for affected adults. Improved access to adult comprehensive SCD care is still a critical need.14
The benefits of adult comprehensive SCD programs include better health outcomes such as fewer acute care visits and hospitalizations. These improvements are anticipated to improve quality of life and result in greater cost-effectiveness as compared with episodic, emergency department (ED)-based care.15-18 Patients with SCD treated at SCD centers use ED and inpatient facilities less frequently, have decreased health care costs overall,15,16,19 and are more likely to be prescribed hydroxyurea,20 which has been demonstrated to improve psychosocial outcomes including health-related quality of life.21
Comprehensive care of SCD requires that affected individuals have a continual bidirectional relationship with their medical team. However, many patients are living in areas in which SCD-focused providers are unavailable (Figure 1). As a result, internists and primary care physicians (PCPs) have been left to provide the majority of SCD care in an acute pain-based or episodic illness-based model. These providers may have limited experience in the care of individuals with SCD and may not keep abreast of SCD guidelines.22 In a survey of family physicians in the United States and Canada, only 20% said they feel comfortable treating patients with SCD.23,24 Thus, without a comprehensive center, affected adults rely on acute care services, often resulting in suboptimal clinical outcomes and extracting a heavy psychological price on the individual.12

Estimated number of individuals with SCD, based on state-specific African American and Hispanic birth-cohort disease prevalence and 2008 US census population, corrected for early mortality. Reprinted from Hassell49 with permission from Elsevier.
Estimated number of individuals with SCD, based on state-specific African American and Hispanic birth-cohort disease prevalence and 2008 US census population, corrected for early mortality. Reprinted from Hassell49 with permission from Elsevier.
Models of comprehensive, team-based management focused on care coordination have demonstrated efficacy in other inherited conditions.25,26 New medications for SCD (eg, crizanlizumab and voxelotor) offer hope for improved outcomes. These medications are promising, but the benefits of any new SCD therapies will not be fully recognized in places where health care access is highly fragmented and specialist care is lacking. Furthermore, these novel medications will not supplant the need for regular comprehensive care.27 Even implementation of current therapies (hydroxyurea, iron chelation) has been limited by lack of access to specialized care.
In 2016, the American Society of Hematology (ASH) launched the Sickle Cell Disease Initiative as their first disease-specific initiative to improve outcomes for people living with SCD in the United States and globally. Elements of this multifaceted initiative include the development of clinical practice guidelines, the launch of a clinical trials network, and the development of a research data hub to capture real-world evidence on patient care. ASH has also complemented these efforts with programs to increase access to care, including advocacy for payment reform and increased provider education.
Specifically recognizing deficiencies in the provision of health care services for adults with SCD, ASH launched an effort to increase the number of adult SCD care programs. Cochairs of this initiative were selected based on their demonstrated success in developing SCD centers and interest in improving access to care. Goals of this effort include increased access for adults to SCD-specific services (infusion centers, appropriate care in ED, etc), transition from pediatric to adult care, and access to the other care professionals needed to address the medical and social aspects of living with SCD.
Methods
To define the most important elements of an SCD center, a convenience sample of 14 established adult SCD treatment centers in the United States and the United Kingdom was sent, via e-mail, a qualitative survey with several questions. The survey was directed toward the leading physician(s) in each center. The first questions asked that each center be defined, including the patient population, staffing, clinic structure, care provision, and outreach efforts, followed by a list of components for assignment into tiers as either (1) essential elements of a sickle cell center (components needed to define themselves as a “sickle cell center”), (2) optimal (but not necessary/essential), or (3) those elements that could be considered suggested (services that improve care delivery but are not essential to provide guideline-based care). Importantly, participants were not asked to include information on their centers’ research capacity or relationship with outside CBOs. The focus of the survey was specific to patient care delivery as an SCD center.
Recognizing that the United States is just 1 of many countries treating SCD, inclusion of non-US–based systems was considered important. However, to be consistent with the goals of the survey, only Canadian and UK clinics were included as they have payer systems that seek to optimize cost-effectiveness similar to the United States. Although their health systems are 1-payer systems, they were felt to be more similar to the United States than the French or Italian payer systems.
The questions included in the survey and the list of potential components of sickle cell centers were composed by the ASH workshop cochairs as we created the workshop. In addition, we considered the requirements of SCD centers per available regulations, including NHLBI SCD guidelines, the UK Forum on Quality Services for Health Services for People with Haemoglobin Disorders, and the Joint Accreditation Commission for Health Care Organizations (JACHO).28,29 Finally, we reviewed the essential qualities of a hemophilia treatment center and included those concepts in the models of care.30 Components listed for categorization included: sickle cell specialist, social worker, case manager, dedicated nurse/nursing staff, advanced care providers, mental health specialist (psychologist or other), PCP, physical therapist, pharmacist, dedicated clinic space or location, infusion center/day hospital/observation unit, and defined business plan. Respondents could also add other components not included in the list that they felt were needed/necessary.
Consensus was defined as at least 75% of respondents agreeing to the placement of components as essential, optimal, or suggested. If a component was only recommended by 1 or 2 physicians, it was immediately placed in the “suggested” category. If there were components that were between categories, another e-mail was sent to ask for sites to choose 1 or the other categories in order to achieve consensus.
Elements that all of the centers classified as “essential” reached consensus easily among center physicians. Other elements that did not reach majority were categorized as “optimal” or “suggested” as described earlier in “Methods.”
Results
Consensus-defined essential components of an SCD center
Comprehensive SCD care requires a programmatic, multidisciplinary, team-based approach. Survey respondents unanimously agreed that all adult SCD centers must offer comprehensive, evidence-based care for adults with SCD that is coordinated throughout the institution. The SCD center should also be the recognized authority (leader within their larger hospital or academic center) for managing SCD as a patient population within their institution/organization. The required and suggested center elements were defined as described herein by the survey participants (Figure 2).
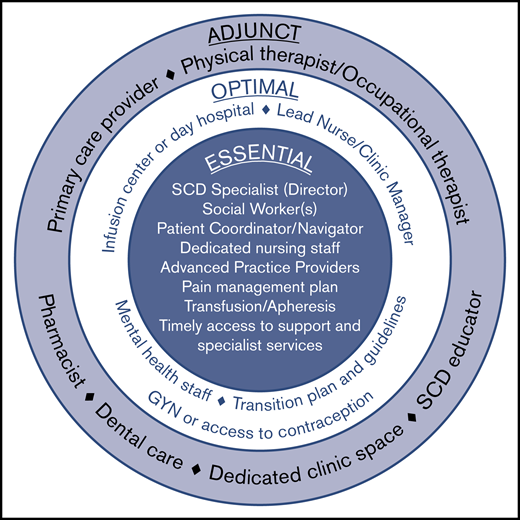
Elements of an adult sickle cell center. Required elements represent the minimal components necessary for a functional and effective comprehensive adult sickle cell center. Optimal elements reflect valuable resources that may be included as part of an adult sickle cell center, although they are not essential. Adjunct elements serve as a supplement to the required and optimal elements and are considered preferred but not strictly necessary.
Elements of an adult sickle cell center. Required elements represent the minimal components necessary for a functional and effective comprehensive adult sickle cell center. Optimal elements reflect valuable resources that may be included as part of an adult sickle cell center, although they are not essential. Adjunct elements serve as a supplement to the required and optimal elements and are considered preferred but not strictly necessary.
At a minimum, an adult SCD center requires a physician lead who is considered a specialist in the care of individuals living with SCD. This individual will accept responsibility for establishing protocols for care, training, implementing audits, and sharing in the overall responsibility for the management of the clinic. The lead SCD specialist should be comfortable providing evidence-based pain management, should undertake continuing professional development of relevance to this role, and have an established plan for how to cover for absences.
Other required elements include 1 or more social workers, a patient coordinator (sometimes called a patient navigator or case manager), and dedicated nursing staff, as well as the ability to offer acute and chronic pain management, transfusions (including apheresis), and timely access to specialist services.
Optimal elements that a center may include are a lead nurse/clinic manager who could also serve as the center director or codirector with the SCD specialist. This person helps share responsibility with the director for upkeep and implementation of guidelines and protocols as well as training and audits related to the center. The clinic manager can have additional responsibilities including quality improvement and oversight.
Although not essential for sickle cell centers, having dedicated infusion space or a day hospital is optimal. Multiple studies have demonstrated the utility of a day hospital to decrease ED care and provide SCD-specific care in a timely manner (especially during pain crisis).31,32 Sickle cell centers also need readily accessible behavioral health staff. Although those surveyed did not feel that having a dedicated psychologist was a “must,” there was universal agreement that it is preferred. Optimal elements also include a transition plan for helping children adapt and transfer to the adult care setting, and available access to a gynecology provider or other means of providing contraception for women with SCD.
In addition to these elements, several other components are considered adjunct (preferred but not strictly necessary). These include a primary care provider (in an embedded model), a physical and occupational therapist, a pharmacist, accessible dental care (as may be available hemophilia treatment centers), and an SCD educator. Also considered adjunct but preferred is a dedicated clinical space and staff that are not shared with other patient populations.
Models of SCD adult care
The centers surveyed were also categorized into different models defined based on the size of the center, patient population, structure (within a division or department), services provided (whether they had primary care within the SCD center), and staffing for care delivery (Figure 3). These models were also defined by their location of services and whether they are stand-alone clinics or embedded within other divisions/departments/clinics.
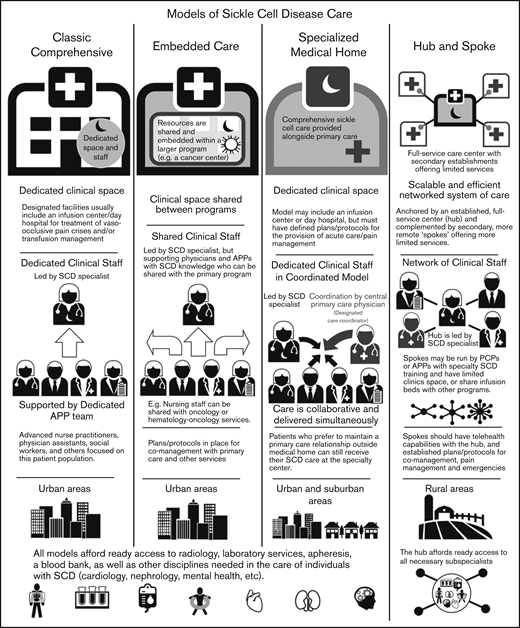
Overview of adult SCD care models. The 4 models of adult SCD care differ with respect to the clinical space and resources, staffing of sickle cell specialists and APPs, the developed environments to which the model is most suited, and other similar factors.
Overview of adult SCD care models. The 4 models of adult SCD care differ with respect to the clinical space and resources, staffing of sickle cell specialists and APPs, the developed environments to which the model is most suited, and other similar factors.
Classic comprehensive care model.
The classic comprehensive care model provides coordinated, team-based SCD care using dedicated clinical space and staff. This type of center is more likely found in urban locations within larger hospital systems or academic centers. These centers usually provide care to a local, relatively large urban SCD population.
A comprehensive clinic will be led by a sickle cell specialist who is supported by a team of advanced practice providers (APPs): advanced nurse practitioners or physician assistants, a social worker, and others focused on this patient population. Facilities usually include an infusion center/day hospital to provide acute care and/or transfusion management. Centers in which an infusion center is not available have prespecified individualized care plans and designated treatment locations for acute management.
By definition, these centers must have ready access to radiology, laboratory services, apheresis, and a blood bank, as well as other disciplines needed in the care of individuals with SCD (Table 1). Most will develop plans and protocols for comanagement with PCPs and other services with a focus on providing team-based, care coordination.
SCD complications and specialists needed
| Complication | Specialists | ES or P |
|---|---|---|
| Retinopathy | Retinal specialist | ES |
| Leg ulcer | Wound care | ES |
| Restrictive lung disease | Pulmonary | ES |
| Pulmonary hypertension | Cardiology (or pulmonary)* | ES |
| Renal disease | Nephrology | ES |
| AVN | Orthopedics | ES |
| Gallstones/hypersplenism | General surgeon | ES |
| Mood disorders | Psychiatry/psychology | ES |
| Neurovascular disease | Neurosurgeon | ES |
| Priapism | Urology | ES |
| Transfusion-related complications | Blood bank specialist | ES |
| Pregnancy-related complications | Maternal-fetal medicine/high-risk OBGYN | ES |
| Iron overload (assessment) | Radiologic specialists | P |
| Chronic pain | Pain specialist | P |
| Complication | Specialists | ES or P |
|---|---|---|
| Retinopathy | Retinal specialist | ES |
| Leg ulcer | Wound care | ES |
| Restrictive lung disease | Pulmonary | ES |
| Pulmonary hypertension | Cardiology (or pulmonary)* | ES |
| Renal disease | Nephrology | ES |
| AVN | Orthopedics | ES |
| Gallstones/hypersplenism | General surgeon | ES |
| Mood disorders | Psychiatry/psychology | ES |
| Neurovascular disease | Neurosurgeon | ES |
| Priapism | Urology | ES |
| Transfusion-related complications | Blood bank specialist | ES |
| Pregnancy-related complications | Maternal-fetal medicine/high-risk OBGYN | ES |
| Iron overload (assessment) | Radiologic specialists | P |
| Chronic pain | Pain specialist | P |
AVN, avascular necrosis; ES, essential; OBGYN, obstetrics and gynecology specialist; P, preferred.
Prefer (not essential) with sickle cell–specific training.
Example.
The Sickle Cell Center for Adults at Johns Hopkins Medicine provides comprehensive services for persons with SCD who live in the greater metropolitan Baltimore, MD and Washington, DC areas. The center offers regularly scheduled outpatient visits, hydroxyurea and transfusion management, genetic counseling, pain management, education, and referral to subspecialty and social services. The center employs full-time hematologists, nurses, APPs, and staff devoted to the care of persons with SCD. Importantly, the center is well embedded within the larger Johns Hopkins Medicine community and staff have established relationships with other departments, notably the ED, which facilitates timely assessments of patients with acute needs. The dedicated clinic space also includes a day hospital for acute pain management.
The infusion center at Johns Hopkins has significantly reduced the admission rate from their ED and 30-day readmissions.15 Community-level impact has also been noted, with a statistically significant 7% annual decrease in the likelihood of readmissions for hospitals in the city of Baltimore.
Embedded care model.
In institutions serving fewer patients with SCD or without enough dedicated space and staff, an embedded SCD care center can provision care by “embedding” itself within a larger, more financially feasible care program and sharing resources. Given the abundance of comprehensive cancer centers and the synergies and similarities between operational infrastructure, several adult SCD programs have formally embedded themselves within hospital-based cancer centers.
In an embedded center, clinical space can be shared between programs, improving efficiency and reducing costs. Personnel can also be shared provided that coordinated, team-based care of SCD patients, who are likely to be far fewer in number than the other patients, can be ensured. These programs still require the leadership of a sickle cell specialist, but supporting physicians and APPs can be shared with the primary program.
In the case of a shared benign hematology/heme-onc model, nursing staff can be shared with hematology-oncology services. Physicians who have sufficient knowledge of SCD management can assist in patient care and provide support to the program. As with the classic comprehensive SCD center model, these centers must offer access to radiology, laboratory services, apheresis, and a blood bank, along with other disciplines needed in the care of individuals with SCD. (Table 1). Embedded centers should also have plans and protocols for comanagement with primary care and other services with a focus on providing team-based, patient-centered care.
Example.
A retrospective analysis of a new adult SCD center embedded within an existing cancer center at the University of Connecticut Health Center demonstrated that this strategy increased the number of annual outpatient preventative visits for chronic disease management, reduced hospitalization rates and length of stay, and decreased hospitalization for management of acute pain.33 Hydroxyurea use among eligible patients increased from 30% before the establishment of the embedded center to 90% after 5 years, indicating that access to dedicated specialist care was likely an important barrier for provision of established treatments. Importantly, the sickle cell program became a regional referral center for adults with SCD. This program was ultimately able to garner substantial hospital resources to transition into a classic comprehensive care model with dedicated space and staffing.
Specialized medical home model.
The medical home concept is a primary care construct traditionally defined as a center for providing patient-centered, team-based, accessible comprehensive care with a focus on quality and safety. The medical home model of care was designed to meet the complexities of chronic disease with care coordination going through a central PCP who acts as a team lead or quarterback.34 Evidence of the benefits of the medical home on quality and costs have been mixed.35
The defining characteristic of a sickle cell–specialized medical home is the colocation of primary care within the comprehensive sickle cell care structure. Like a classic comprehensive center, a SCD specialized medical home will have dedicated clinical staff and space, along with a designated care coordinator. In this setting, the lead sickle cell specialist acts as the team leader but the care is collaborative and delivered simultaneously. This model may or may not include an infusion center or day hospital, but must have defined plans for the provision of acute care/pain management. This model can be considered in both urban and suburban settings, but is unlikely to have enough patients in its catchment area for financial feasibility in rural locations. However, when patients present from farther afield, the opportunity to have all of their medical needs accommodated in 1 place is of value. Patients who prefer to maintain a PCP relationship outside of the medical home can still receive SCD care at the specialty center. As with the other models of care, these centers must have access to radiology, laboratory services, apheresis, a blood bank, and other services needed for the care of individuals with SCD (Table 1).
Example.
The Lifespan Comprehensive Sickle Cell Center at the Medical University of South Carolina (MUSC) provided both specialized sickle cell care and primary care for affected adults from 2014 to 2018. Patients seen at the center were able to see both providers at each visit; an attached infusion center allowed for acute care visits, including transfusion management and pain medications for acute crises.
Over 400 adults with SCD were followed longitudinally by the Lifespan center at MUSC. Just over one-half of patients (55%) opted to receive their primary care at the center, primarily for ease of scheduling and coordinated communication. At program entry, the majority of these individuals had not seen a PCP in the previous 12 months (J.K., unpublished observational data, MUSC years 2014-2019). For the 45% of patients who preferred to continue seeing their “outside” PCP, only SCD-specific care was provided at Lifespan. To assess the effectiveness of the program, a group of 27 Medicare-identified accountable care organization patients were identified retrospectively. Only those followed consistently from 2015 to 2017 were included in the analysis. These individuals saw a 65% decrease in ED visits and a 46.5% decrease in hospitalizations (J.K., unpublished observational data, MUSC years 2014-2019).
Another example of a specialized medical home model is The Adult Comprehensive Sickle Cell Center at The Ohio State University (Columbus, OH). This program has partnered with the internal medicine physicians at the medical center to provide in-house–based primary care to over 40 patients. The internal medicine provider visits the patient at home to allow for longer, more comprehensive evaluations of the patients’ health and needs. In conjunction with their SCD care within the comprehensive center, these individuals received their primary care. In addition to improved SCD outcomes, this program has demonstrated improvements in primary care outcomes, including immunization rates and cancer-screening rates. Additionally, since the program started, admissions, readmissions, and total number of hospital days have decreased, along with a modest decrease in ED utilization.
Hub-and-spoke model.
Individuals with SCD living in rural areas are more likely than their urban counterparts to lack comprehensive coordinated care. These patients have higher rates of acute care utilization and readmission,36 and greater dissatisfaction with the care received, often related to the longer wait times in rural EDs.37 Even living farther from an established urban sickle center, but still within its catchment area, can adversely impact care.38,39 Telemedicine and telementoring have been proposed as ways of extending care from urban centers to rural regions.40,41
The term “hub and spoke” offers a vivid image of how this model can expand access to care. The hub-and-spoke design provides a networked system of care anchored by an established, full-service care center (hub) complemented by secondary establishments (spokes) that offer more limited services, but alleviate acute shortages in care. As an organizational design, hub and spoke is readily scalable and can operate efficiently with the replication of operations across multiple sites.42 A basic example is a health care network that consists of a main campus and 1 or more satellite campuses.
Centers designed according to any of the care models mentioned (the classic comprehensive, the embedded, or the specialized medical home) could serve as the central hub from which spokes extend to provide care to geographically distant areas. As previous discussed, the hub will be led by a sickle cell specialist and have ready access to all necessary subspecialists. Spokes may be run by PCPs with contacts to the hub or by APPs with specialty training in SCD management. These spokes may have dedicated infusion areas, but are more likely to share infusion space. Spokes must be able to provide transfusion and infusion services on the same campus. Spokes should also have telehealth capabilities with the hub, and established protocols for comanagement and the management of acute pain and emergencies. An advantage of this model is the potential to extend care to areas remote from a larger SCD care center at lower cost.
Example.
The Lifespan Comprehensive Sickle Cell Clinic at MUSC serves as a “hub” for the hub-and-spokes state network, called (SC).2 The (SC)2 network includes spokes throughout South Carolina in Beaufort, Columbia, and Georgetown. The Beaufort Memorial Hospital (BMH) is 1 of the spokes of the network. The SCD clinic at BMH is run by a full-time APP (a nurse practitioner) who trained with the hub prior to the opening of the spoke. The spoke clinic provides care Monday through Friday and serves 40 individuals with SCD in the Beaufort area. The clinic offers transfusion therapy, hydroxyurea management, and acute pain management with individualized care plans, and has local, emergency backup from the ED and the hospitalist service. This spoke clinic provides care under the supervision of the “hub” provider, checking in as needed on patient care issues. The supervising SCD provider from the hub uses telemedicine to see patients remotely, both on a scheduled and as-needed basis, and travels to assess patients at their home spoke clinics on a quarterly basis. These in-person clinics also provide the SCD specialist provider an opportunity to assess new patients.
Economic aspects of an SCD center
Improving access to regular and expert care for individuals with SCD can be a financially advantageous strategy. Patients who lack comprehensive management require more acute hospital care, which is more expensive than the provision of comprehensive, regular care.15,19 The charges associated with SCD care are disproportionate to its prevalence and complexity. In 2006, there were an estimated 188 194 ED visits among adults and 44 188 ED visits among children in the United States, with total charges estimated to be $356 million dollars.43 Combined ED and inpatient charges were estimated to be $2.4 billion dollars. Individuals with SCD had 3 times the charges for hospitalization associated with an ED visit (per 100 patients with the disease) compared with those with congestive heart failure.43
Individuals treated in dedicated sickle cell day hospitals have fewer ED visits, lower admission rates, and shorter hospital stays; therefore, the potential exists for substantial overall savings for the institution and the payer.15,31,44,45 Cost savings must be viewed from several perspectives: patient, hospital system, and the payer. The data support the premise that access to SCD-specific care and outpatient infusion results in a decrease in hospital admissions. Although the total cost differential is dependent on length of stay, procedures, and tests ordered, etc, the decrease in hospital admissions translates into overall less money spent on hospital-related charges regardless of who is paying the bill.46 In 1 study, 3 years after opening a sickle cell unit, a 43% decrease in hospital admissions was demonstrated along with a 49% decrease in occupied bed days.44 In the third year of operation, 84% of patients treated for pain episodes were managed without the need for hospital admission. Although it is not clear that the hospital system did not lose money that would have been reimbursed to them, it is likely, based on other similar reports,47,48 that the cost, including subsequent readmission costs, would have exceed the payer-based diagnosis-related group funding, resulting in a loss for the institution.
The ASH SCD Centers Workshop
In November 2019, ASH hosted its inaugural SCD Centers Workshop, a 3-day in-person event held at ASH headquarters in Washington, DC. The workshop was led by 4 cochairs who are all directors of adult sickle cell treatment programs and who have extensive experience in developing comprehensive adult treatment centers. Additional faculty included those with backgrounds in financial analysis, social work, and care coordination.
Applicants included clinical interprofessional teams seeking to develop adult comprehensive care programs. Members of the team included a clinical medical champion for the center (usually a hematologist or general internist, but possibly an APP), business officials from the institution, and additional interested health care professionals.
Personnel from 11 institutions with nascent adult SCD programs attended. The curriculum included didactics and small group breakout sessions on a number of (nonclinical) requirements for a SCD center, including how to obtain “buy-in” and financial support from a health care institution, collaboration with SCD stakeholders and their support organizations, and team building and appropriate training for staff, including how to address potential unconscious bias.
The workshop focused on explaining the core elements of a center, as noted herein, and the ways in which care for affected adults can be optimized. Attendees were assigned to a faculty member/mentor from within their geographic region. Mentors and attendees met multiple times throughout the 3-day workshop to identify the optimal model for their center, strategize methods for refining their team, and to begin writing their business plan. Over the next 12 months, attendees will continue to be mentored in establishing sickle cell centers.
Discussion
The designation of an SCD comprehensive center will create the ability to standardize the implementation of SCD guidelines and assess their impact on patient outcomes. These centers can also work together to identify areas in which evidence is lacking. Importantly, the infrastructure of center-based care will allow for enhanced comparative quality and safety metrics as well as cooperative, postapproval assessments of novel therapies. Center designation will also codify, legitimize, and promote comprehensive sickle cell centers within their current hospital/academic environment and enhance the careers of current and future center directors and staff (both locally and nationally).
Several features of currently established centers have not been included in this description, notably the capacity for doing clinical research and the need for relationships with sickle cell-focused CBOs. The present project seeks to define and maintain centers based on their ability to deliver quality, team-based, patient-centered care that is economically sustainable. Although the hope is that both new and established SCD centers will participate in clinical research and engage with local CBOs, it is not considered a requirement.
In conclusion, comprehensive care for individuals living with SCD is a necessity. Essential elements are required for an SCD center to provide team-based care. However, the model of care used can vary according to location, personnel, the size of the population served, and financial and institutional considerations. SCD centers for adults must provide compassionate, comprehensive care that is coordinated to ensure continuous and personalized care with attention to both the physical and emotional well-being of the individual. Providing standardized, equitable patient-centered management will ensure higher quality, more cost-effective care for this vulnerable patient population.
Protocols and information related to this article may be obtained through e-mails to the corresponding author, Julie Kanter (jkanter@uabmc.edu).
Acknowledgment
The authors thank the American Society of Hematology (especially Charles Clayton) for continued support in this effort.
Authorship
Contribution: J.K. designed the study, conducted the qualitative interviews with participating sickle cell centers, and wrote the first draft of the article; J.K. and S.L. helped to review the findings and develop the care models; J.K., W.R.S., J.D.R., and S.L. were cochairs of the SCD Centers Workshop; and all authors critically reviewed and approved the final version of the manuscript.
Conflict-of-interest disclosure: J.K., S.L., W.R.S., P.C.D., J.D.R., and S.C. received travel support and an honorarium from ASH for serving as faculty for the SCD workshop. J.K. has served on scientific advisory boards for Novartis Pharmaceuticals, Global Blood Therapeutics, Bluebird Bio, and Editas; has received honorariums for travel and speaking opportunities from Terumo, Rockpointe, and Medscape; serves on steering committees for Novartis and AstraZeneca; is site principal investigator on studies funded by Bluebird Bio, Novartis, Shire, and Imara; and reports consulting from Novartis. W.R.S. reports being a consultant for Emmaus Pharmaceuticals, Global Blood Therapeutics, Novartis, and Pfizer, and an investigator for Emmaus Pharmaceuticals, Global Blood Therapeutics, Health Resources and Services Administration (HRSA), NHLBI, National Institutes of Health, Novartis Pharmaceuticals, Patient-Centered Outcomes Research Institute (PCORI), Pfizer, Inc, Baxalta-Shire, and Imara. M.T. reports being a consultant for Pfizer, Inc and Ironwood Pharmaceuticals. B.A. reports research funding from Imara and Novartis and consulting for Bluebird Bio, CRISPR, Cyclerion, GBT, Hemanext, Novartis, NovoNordisk, Terumo BCT, and Vertex. P.C.D. reports being a consultant for Pfizer, GBT, Novartis, Ironwood Pharma; and research funding from National Institutes of Health, University of Pittsburgh. J.L. received research support from GBT and Bluebird Bio, and was an advisor to GBT × 1 in 2019. J.J.S. reported institutional research funding from Takeda to support a clinical trial of rADAMTS13 for the treatment of SCD. D.G.M. reports research support from GBT, BBB, and Grifols, and consulting for Novartis, Pfizer, GBT, and Forma Therapeutics. I.O. reported consultancy for Novartis, Pfizer, Cyclerion, and FORMA Therapeutics; speakers’ bureaus for Novartis, Terumo, and GBT; advisory board participation for Novartis, Pfizer, GBT, Acceleron, Cyclerion, and FORMA Therapeutics; being a site principal investigator on studies funded by Novartis, GBT, and Cyclerion; receiving grants from HRSA, PCORI, North Carolina Division of Public Health (NC DPH), and Hemedicus (educational); data and safety monitoring board membership for Micella Biopharma; and being Editor-In Chief for Hematology News. J.D.R. is a consultant for Truven Health Analytics and Community Health Network of Connecticut. S.L. was site principal investigator on studies funded by Novartis, Pfizer, GBT, Ironwood, and Shire. The remaining authors declare no competing financial interests.
Correspondence: Julie Kanter, University of Alabama Birmingham, 1720 2nd St South, NP 2510, Birmingham, AL 35233; e-mail: jkanter@uabmc.edu.

