

Key Points
JAK2S523L is a novel activating JAK2 mutation in JAK2V617F− MPNs.
JAK2S523L occurs in a residue that is critical for the negative regulation of JAK2 kinase activity.

Abstract
The SH2-JH2 linker domain of JAK2 has been implicated in the negative regulation of JAK2 activity. In 2 patients with myeloproliferative neoplasms (MPNs), we identified and characterized the novel JAK2 mutation S523L, which occurs in a key residue in the linker region. In 1 case, acquisition of JAK2S523L was associated with thrombocytosis and bone marrow megakaryocytic hyperplasia, and there were no other somatic alterations in this patient. The second patient with JAK2S523Lmutation presented with increased hematocrit and had concurrent mutations in RUNX1 and BCORL1. Consistent with the genetic and clinical data, expression of JAK2S523L causes interleukin-3–independent growth in Ba/F3 cells transduced with the erythropoietin receptor by constitutively active Jak2/Stat5 signaling.
Introduction
Myeloproliferative neoplasms (MPNs) are clonal disorders of hematopoietic stem/progenitor cells that manifest with an increased number of mature myeloid cells. The most common BCR-ABL1− MPNs are polycythemia vera (PV), essential thrombocythemia, and primary myelofibrosis. The JAK2V617F mutation1-4 is identified in ~98% of PV, 35% to 57% of primary myelofibrosis, and 23% to 57% of essential thrombocythemia patients.5-7 It occurs in the JH2 pseudokinase autoinhibitory domain and leads to constitutive JAK2 activation, increased sensitivity to cytokine signaling, and erythrocytosis in preclinical models.8 Molecular screening of PV patients lacking the JAK2V617F mutation has identified >10 other JAK2 mutations in exon 12, a majority within residues 536 to 5448 (eg, N542-E543 del [23%], E543-D544del [11%], and F537-K539delinsL and K539L [10%]).9,10 These mutations are most commonly characterized by isolated erythrocytosis and have been shown to confer a proliferative advantage and increased downstream signaling of JAK2 in the absence of cytokines.8,10
The tyrosine kinase activity of JAK2 is tightly regulated. The intramolecular cis interaction between the JH2 pseudokinase and the JH1 tyrosine kinase domain of JAK2 negatively regulates JH1 tyrosine kinase activity and maintains the kinase in an inactive state in the absence of cytokine stimulation.11-13 Phosphorylation of Y570 in the JH2 domain and S523 in the SH2-JH2 linker domain has been shown to interact with the JH1 domain and enforce the JH2-JH1 autoinhibitory interaction.14-17 These data suggest mutations at the JH2-JH1 interface, either in the JH2 domain (eg, R683G, L611S) or in the JH1 domain (D873N, P933R), or mutations at the negative regulatory sites Y570 or S523 destabilize the JH2-JH1 interaction and enhance JAK2 signaling. Here we report clinical and molecular data of 2 patients who presented with JAK2V617F− MPNs with novel mutations at serine 523, specifically the S523L mutation, which we describe and functionally characterize.
Methods
Sequencing
Peripheral blood samples were subjected to Sanger sequencing of JAK2 exon 12 and next-generation sequencing of 54 myeloid neoplasm–associated genes (TruSight Myeloid Sequencing Panel; Illumina), including JAK2 exon 12. Mutational analysis of MPL and CALR was performed using Sanger sequencing. The BCR-ABL1 fusion transcript was ruled out by reverse transcription polymerase chain reaction.
In vitro mutagenesis
Jak2S523Land Jak2K539L mutations were introduced into murine Jak2 wild-type (WT) MSCV-IRES-GFP using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Affymetrix) with the following primers: Jak2S523L, F-mutagenesis: aaatggtatttctgatgttcagatcttaccaacattacagaggc and R-mutagenesis: gcctctgtaatgttggtaagatctgaacatcagaaataccattt and Jak2K539L, F-mutagenesis: ttcattaaatattaaatcttcattcctgattaagtgaaacaccatttgattcacattattatgc and R-mutagenesis: gcataataatgtgaatcaaatggtgtttcacttaatcaggaatgaagatttaatatttaatgaa.
IL-3 withdrawal
Ba/F3 cells stably expressing the murine erythropoietin receptor (EPOR) (Ba/F3-EPOR)18 or the human MPL (Ba/F3-MPL) were grown in RPMI medium (10% fetal calf serum, penicillin/streptomycin) and transduced with retroviral supernatant containing MSCV-Jak2WT-GFP, MSCV-Jak2V617F-GFP, MSCV-Jak2S523L-GFP, or MSCV-Jak2K539L-GFP, respectively. Flow-sorted green fluorescent protein (GFP)–positive Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L cells coexpressing either EPOR or MPL were plated (6-well plates; 106 cells per 3 mL per well) in interleukin-3 (IL-3)–free RPMI (10% fetal calf serum, penicillin/streptomycin). Cell numbers were counted using the Beckman Coulter ViCell XR cell counter.
WB and immunoprecipitation
Transduced Ba/F3-EPOR and Ba/F3-MPL cells were grown in the presence (Jak2 WT) or absence (Jak2V617F, Jak2S523L, or Jak2K539L) of IL-3 for 4 to 6 hours. For assessment of effects of ruxolitinib on signaling pathways, cells were grown in the absence or presence of ruxolitinib (1 µM) for 4 hours before lysis. Lysate (30-40 µg) was separated on 4% to 12% Bis-Tris electrophoresis gels and probed for Jak2 (Cell Signaling Technologies [CST] #3230), phosphorylated Jak2 (pJak2; CST #3776), Stat5 (CST #9363), pStat5 (CST #9351), Akt (CST #4691), pAkt (CST #4060), Erk1/2 (CST #9102), pErk1/2 (CST #9101), and Cofilin (CST #5175). Jak2 was immunoprecipitated from 600 to 900 µg of lysate followed by western blot (WB) with anti-pS523 and anti-pY570, provided by Martin G. Myers Jr.
In vitro proliferation assay during ruxolitinib treatment
For proliferation assays, 100 000 cells per 200 µL of medium were plated in triplicate and supplemented with increasing doses of ruxolitinib using 9-point, threefold dilutions with a top concentration of 100 µM. After 48 hours, proliferation was assessed using the CellTiter-Glo Luminescent Cell Viability Assay (Promega) and normalized to proliferation in media with an equivalent volume of dimethyl sulfoxide. Results were illustrated using GraphPad Prism 8.0.
Results
The first patient was a 48-year-old man who presented with an increased hemoglobin level (18 g/dL) and hematocrit (53%) while being followed for hypertension. White blood cell count was 9.2 × 109/L and platelet count was 319 × 109/L at the time of diagnosis. The second patient was a 36-year-old woman who presented with an increased platelet count; her platelet counts had been in the range of 534 × 109/L to 701 × 109/L over a 15-year period. A bone marrow biopsy at the time of initial diagnosis showed megakaryocyte hyperplasia without other abnormalities. Cytogenetic analysis was normal.
JAK2V617F, MPL, and CALR mutations were not detected (Figure 1A). Screening for JAK2 exon 12 mutations detected point mutation c.1568C>T, leading to an amino acid change from serine to leucine in peripheral blood samples from both patients, but not in matched germ line DNA (Figure 1B). Next-generation sequencing confirmed somatic JAK2S523L mutations in both patients (Figure 1C) and revealed missense mutations of RUNX1 (G69R) and BCORL1 (P810L) in the first patient. No additional mutations were detected in the second patient. The JAK2S523L mutation is localized in the linker region between the SH2 and pseudokinase (JH2) domains of JAK2 (Figure 1D).

Detection of the JAK2S523L mutation in 2 patients. (A) Clinical features and cooccurring molecular alterations detected by Sanger and next-generation sequencing (NGS). (B) Conventional Sanger sequencing in peripheral blood and buccal swabs reveals a somatic point mutation at c.1568, which causes an amino acid change from TCA to TTA. (C) Detection of the JAK2S523L mutation by NGS of 54 myeloid neoplasm–associated genes using a targeted panel (TruSight Myeloid Sequencing Panel; Illumina). (D) Schematic illustration of JAK2 structure and the localization of the JAK2S523L mutation. BM, bone marrow; Hb, hemoglobin; Hct, hematocrit; NA, not applicable; plts, platelets; VAF, variant allele frequency.
Detection of the JAK2S523L mutation in 2 patients. (A) Clinical features and cooccurring molecular alterations detected by Sanger and next-generation sequencing (NGS). (B) Conventional Sanger sequencing in peripheral blood and buccal swabs reveals a somatic point mutation at c.1568, which causes an amino acid change from TCA to TTA. (C) Detection of the JAK2S523L mutation by NGS of 54 myeloid neoplasm–associated genes using a targeted panel (TruSight Myeloid Sequencing Panel; Illumina). (D) Schematic illustration of JAK2 structure and the localization of the JAK2S523L mutation. BM, bone marrow; Hb, hemoglobin; Hct, hematocrit; NA, not applicable; plts, platelets; VAF, variant allele frequency.
We next assessed the functional significance of these mutations in vitro. Given the clinical phenotypes of the 2 patients, with either erythrocytosis or thrombocytosis, we generated Ba/F3 cell lines stably expressing the mutant Jak2S523L (or its controls, Jak2 WT, Jak2V617F, or Jak2K539L) in combination with either the EPOR or thrombopoietin receptor (MPL). Ba/F3 cells stably expressing MPL/Jak2S523L proliferated in the absence of IL-3, similar to the MPL/Jak2V617F and MPL/Jak2K539L cells, whereas Ba/F3-MPL/Jak2 WT cells did not grow in the absence of IL-3 (Figure 2A). In line with these results, in EPOR-overexpressing Ba/F3 cells, expression of Jak2S523L conferred cytokine-independent growth, similar to expression of Jak2V617F or the exon 12 Jak2K539L mutation (Figure 2B).
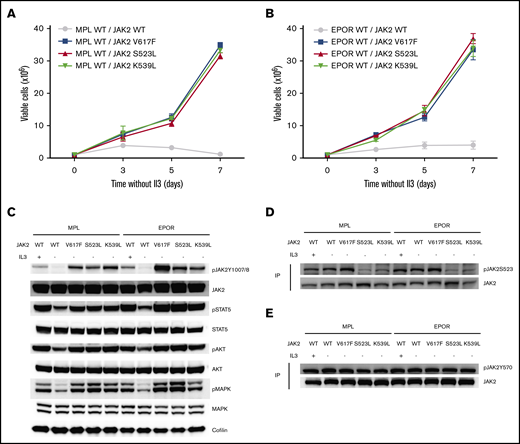
The Jak2S523L mutation causes IL-3–independent growth in Ba/F3 cells expressing EPOR through Jak2/Stat5 activation by impairing phosphorylation of S523, a negative regulatory site for Jak2. (A) Ba/F3 cells transduced with MPL and either Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L were grown in the absence of IL-3. Results of 3 independent experiments are depicted as means ± standard errors of the mean. (B) Ba/F3 cells transduced with EPOR and either Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L were grown in the absence of IL-3. Results of 3 independent experiments are depicted as means ± standard deviations. (C) WB analysis of Jak2/Stat5, Akt and mitogen-activated protein kinase (Mapk) signaling in Ba/F3-MPL or Ba/F3-EPOR cells transduced with Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L. (D-E) Lysates from Ba/F3 cells were immunoprecipitated (IP) with total Jak2 antibody and analyzed by WB using antibodies for pS523 and pY570, respectively.
The Jak2S523L mutation causes IL-3–independent growth in Ba/F3 cells expressing EPOR through Jak2/Stat5 activation by impairing phosphorylation of S523, a negative regulatory site for Jak2. (A) Ba/F3 cells transduced with MPL and either Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L were grown in the absence of IL-3. Results of 3 independent experiments are depicted as means ± standard errors of the mean. (B) Ba/F3 cells transduced with EPOR and either Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L were grown in the absence of IL-3. Results of 3 independent experiments are depicted as means ± standard deviations. (C) WB analysis of Jak2/Stat5, Akt and mitogen-activated protein kinase (Mapk) signaling in Ba/F3-MPL or Ba/F3-EPOR cells transduced with Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L. (D-E) Lysates from Ba/F3 cells were immunoprecipitated (IP) with total Jak2 antibody and analyzed by WB using antibodies for pS523 and pY570, respectively.
WB analyses revealed cytokine-independent activation of the Jak2/Stat5 pathway in Jak2S523L-expressing cells (Figure 2C). Furthermore, activation of the Jak2/Stat5 pathway in Jak2S523L-expressing cells was associated with impaired phosphorylation of S523, a negative regulatory site for Jak2 activity (Figure 2D). The phosphorylation status of Y570, another Jak2 regulatory site, was not significantly altered in Jak2S523L-mutant cells (Figure 2E).
Ba/F3 cells harboring the Jak2S523L mutation in combination with either MPL or EPOR displayed sensitivity to ruxolitinib equal to that displayed by cells expressing Jak2V617F (Figure 3A-B). Ruxolitinib treatment showed similar attenuation of Stat5, Akt, and mitogen-activated protein kinase signaling in cells expressing the Jak2S523L,Jak2V617F, or Jak2K539L mutation (Figure 3C-D) and slightly increased phosphorylation at Jak2 Y1007/1008 but did not affect phosphorylation at Jak2Y570 or Jak2S523 (supplemental Figure 1).
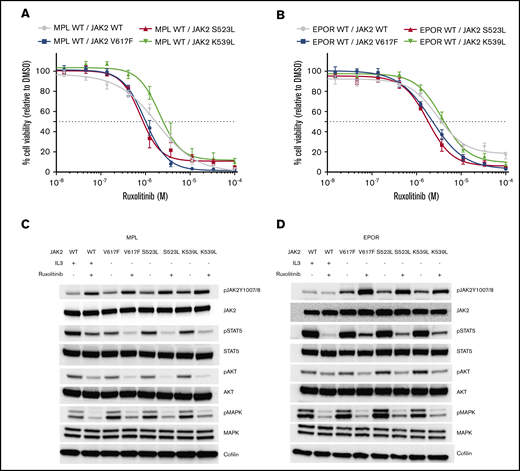
Jak2S523L-expressing Ba/F3 cells coexpressing EPOR or MPL cells are sensitive to treatment with ruxolitinib. (A) Proliferation with increasing concentration of ruxolitinib relative to proliferation in the presence of dimethyl sulfoxide (DMSO) control is depicted for Ba/F3 cells transduced with MPL coexpressing either Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L. Data are depicted as means ± standard errors of the mean (SEMs). (B) Proliferation with increasing concentration of ruxolitinib relative to proliferation in the presence of DMSO control is depicted for Ba/F3 cells transduced with EPOR coexpressing either Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L. Data are given as means ± SEMs. (C) WB analysis. Ruxolitinib treatment for 4 hours at 1 µM inhibits pStat5, pAkt, and phosphorylated mitogen-activated protein kinase (pMapk) signaling in Ba/F3 cells transduced with MPL coexpressing either Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L. (D) WB analysis. Ruxolitinib treatment for 4 hours at 1 µM inhibits pStat5, pAkt, and pMapk signaling in Ba/F3 cells transduced with EPOR coexpressing either Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L.
Jak2S523L-expressing Ba/F3 cells coexpressing EPOR or MPL cells are sensitive to treatment with ruxolitinib. (A) Proliferation with increasing concentration of ruxolitinib relative to proliferation in the presence of dimethyl sulfoxide (DMSO) control is depicted for Ba/F3 cells transduced with MPL coexpressing either Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L. Data are depicted as means ± standard errors of the mean (SEMs). (B) Proliferation with increasing concentration of ruxolitinib relative to proliferation in the presence of DMSO control is depicted for Ba/F3 cells transduced with EPOR coexpressing either Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L. Data are given as means ± SEMs. (C) WB analysis. Ruxolitinib treatment for 4 hours at 1 µM inhibits pStat5, pAkt, and phosphorylated mitogen-activated protein kinase (pMapk) signaling in Ba/F3 cells transduced with MPL coexpressing either Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L. (D) WB analysis. Ruxolitinib treatment for 4 hours at 1 µM inhibits pStat5, pAkt, and pMapk signaling in Ba/F3 cells transduced with EPOR coexpressing either Jak2 WT, Jak2S523L, Jak2V617F, or Jak2K539L.
Discussion
JAK2 is the critical kinase for mediating cellular signaling by type I/II cytokine receptors (eg, EPOR, thrombopoietin receptor, granulocyte-macrophage colony-stimulating factor, or interferon γ receptor). Cytokine binding to their cognate receptors induces dimerization of JAK2s, resulting in auto/transphosphorylation of the activation loop Tyr1007/1008 residues and subsequent phosphorylation of other potentiating residues, such as Tyr637, Tyr813, Tyr868, Tyr966, and Tyr972, as well as phosphorylation of residues that negatively regulate JAK2 activity, such as Tyr119, Tyr221, Tyr317, Tyr570, and Tyr913.14-17,19-21 Ser523 is the only residue that is constitutively phosphorylated in JAK2.16,19
JAK2 consists of FERM and SH2-like domains, a JH2 pseudokinase domain, and a JH1 tyrosine kinase domain. The JAK2 JH2 domain is a mutational hotspot in JAK2 linked to a hyperactive JAK2 and MPN pathophysiology.22 The SH2-JH2 linker domain reinforces the JH2-JH1 autoinhibitory interaction, which plays a role in the negative regulation of JAK2 kinase activity and reduction of JH1 domain affinity for ATP.13,22 As a consequence, mutations in the JH2 and SH2-JH2 linker domains disrupt the autoinhibitory pose and constitutively activate JAK2 signaling.15,22 The JH2 domain has been shown to negatively regulate JH1 activation by allosteric inhibition in the JH1-JH2 autoinhibitory dimer,13 which is reinforced by phosphorylation of Ser523 and Tyr570.19 Phosphorylation of JAK2 at Ser523 and its negative role in the regulation of JAK2 activity were identified by Mazurkiewicz-Munoz et al,17 who introduced a serine-to-alanine mutation at residue 523 and showed that this substitution resulted in enhanced JAK2 tyrosine kinase phosphorylation and increased JAK2/STAT5 signaling.16,23
To our knowledge, this is the first identification of somatic mutations at JAK2S523 in human disease. We demonstrate that mutations at this residue transform Ba/F3 cells, confer cytokine-independent growth, and constitutively activate Jak2/Stat5 signaling. Similarly to the experimentally introduced serine-to-alanine substitution,17 the JAK2S523L mutation leads to a change from a polar amino acid (serine) to a nonpolar, hydrophobic leucine and removes the negative regulatory phosphorylation site. We hypothesize that the abrogated Ser523 phosphorylation then leads to dysregulated JAK2 activation. Taken together, these data demonstrate the pathophysiologic significance of Ser523 mutations in the SH2-JH2 linker domain of JAK2 in the pathogenesis of MPNs and underscore the role of autoinhibitory phosphorylation, including at S523, in regulating JAK2 kinase activation.
Requests for data sharing should be e-mailed to the corresponding author, Ross L. Levine (leviner@mskcc.org).
Acknowledgments
The authors acknowledge Martin G. Myers Jr, who kindly provided anti-pS523 and anti-pY570 antibodies; the patients who consented to use their clinical and sequencing data; and all physicians involved in the care of these patients.
This work was supported by Memorial Sloan Kettering Cancer Center support grant P30 CA008748 from the National Cancer Institute (NCI), National Institutes of Health (NIH), by NCI, NIH, grants R35 CA197594-01A1 and P01 CA108671 11 (R.L.L.), by the Janus Fund (O.S. and R.L.L.), and by German Research Foundation grant PA 2541/1-1 (F.P.).
Authorship
Contribution: B.Y. identified the patients with the JAK2S523L mutation and provided clinical and molecular patient data; F.P. and R.L.L. planned experiments; O.S. and H.M.H. provided the pS523 and pY570 antibodies; F.P. performed the experiments; A.K. contributed to performance of WB; B.Y. and R.L.L. mentored and supervised the work; F.P., R.L.L., B.Y., O.S., and H.M.H. interpreted the data; F.P., B.Y., and R.L.L. wrote the manuscript; and all authors revised and approved the manuscript.
Conflict-of-interest disclosure: O.S. received competitive research funding from the Academy of Finland, Sigrid Jusélius Foundation, Finnish Cancer Foundation, Jane and Aatos Erkko Foundation, Tampere Tuberculosis Foundation, and Pirkanmaa Hospital District. R.L.L. is on the supervisory board of Qiagen and is a scientific advisor to Imago, Mission Bio, Zentalis, C4 Therapeutics, and Isoplexis; receives research support from and consulted for Celgene and Roche and has consulted for Incyte, Janssen, Astellas, Morphosys and Novartis; and has received honoraria from Roche, Lilly, and Amgen for invited lectures and from Gilead for grant reviews. The remaining authors declare no competing financial interests.
Correspondence: Ross L. Levine, Memorial Sloan Kettering Cancer Center, 1275 York Ave, Box 20, New York, NY 10065; e-mail: leviner@mskcc.org; or Benedict Yan, Molecular Diagnosis Centre, National University Hospital, 1E Kent Ridge Rd #13-00, Singapore 119228; e-mail: benedict_yan@nuhs.edu.sg.

