

Key Points
Whole genomic and transcriptomic analyses of MEITL revealed multiple potential therapeutic targets.
Synergistic effects of pimozide and romidepsin are shown in a well-characterized MEITL PDX model.
Introduction
Monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL) is a highly aggressive malignancy with an overall median survival of only 7 months.1 In addition, only 38 patients with MEITL were identified from 4 institutes over a period of 19 years.2 These factors pose great challenges to better understanding and managing the disease. Therefore, there is a dire need to establish clinically relevant models to explore the efficacy of therapeutic targets to improve the management of MEITL.
Methods
Genomic studies
Whole-genome sequencing (WGS) and whole-transcriptomics profiling were performed for 4 MEITL tumors (supplemental Table 1). Whole exome sequencing (WES) was performed for patient-derived xenograft (PDX) tumors. Details are described in the “Materials and methods” in the supplemental Materials. The human subject study was approved by the SingHealth Centralized Institutional Review Board (2004/407/F).
Animal experiments
All experiments were approved by the SingHealth Institutional Animal Care and Use Committee. For the PDX model, fresh tumor tissue was minced and subcutaneously implanted in the flank of NSG mice. For the orthotopic model, minced tissue was intraperitoneally inoculated into the mice. The animal study was approved by the SingHealth Institutional Animal Care and Use Committee (2018/SHS/1423).
Quadratic phenotypic optimization platform
Dissociated cells were treated with drug combinations based on the orthogonal array composite design.3 A total of 12 drugs at 3 concentrations were interrogated, and of a total of more than 500 000 combinations, 155 were empirically tested. The correlation of treatment combination (input) and corresponding cell viability (output) was fitted into a second-order quadratic equation, generating ranked drug combinations and predicted cell viability. The details of the quadratic phenotypic optimization platform (QPOP) are described in previous publications.4,5
The rest of the “Materials and methods” are described in the supplemental Materials.
Results and discussion
Genomic landscape of MEITL
To elucidate the whole-genome profile of MEITL, we performed WGS on 4 MEITL tumors from separate cases and matched normal blood samples with an average depth of 68× and 59× (supplemental Table 2). An average of 1.52 somatic short variants per megabyte was called from each sample. Neither microsatellite instability nor hypermutation was detected. A total of 338 nonsilent protein-coding mutations (average, 84.5; range, 62-121; supplemental Table 3) were detected, and recurrent mutations were validated in CREBBP, STAT5B, SETD2, GNAI2, JAK3, and AXSL3 (Figure 1A), which are consistent with those in previous studies.1,6,7
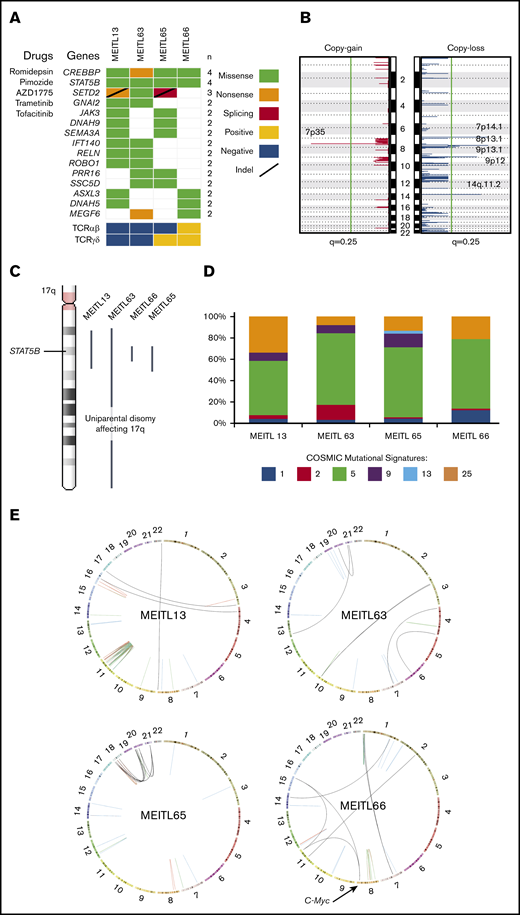
The whole-genome–wide landscape of MEITL. (A) Oncoplot of recurrent somatic short-variant mutations detected in 4 MEITL tumors. Accompanying compounds that can target the corresponding gene mutations are labeled to the left of the oncoplot. (B) Recurrent somatic copy number alteration across the 4 MEITL using WGS data: copy-gain (left) and copy-loss (right) events. The vertical green line denotes the q = 0.25 (adjusted P value) cutoff for significant copy number events. (C) 17q arm-wide shows that large-scale, copy-neutral, loss-of-heterozygosity alterations were present and affected the STAT5B loci in all our MEITLs. (D) The 100% stacked bar plots comparing the proportions of known COSMIC (Catalogue of Somatic Mutations in Cancer) mutational signatures within each MEITL tumor. The denoted mutational signature numbers are as described at https://cancer.sanger.ac.uk/cosmic/signatures. (E) Circos (http://mkweb.bcgsc.ca/circos) plots depicting the structural rearrangements as links for each WGS sample. Links are colored differently for each type of structural rearrangement: red for duplications, blue for deletions, green for inversions, and black for interchromosomal translocations.
The whole-genome–wide landscape of MEITL. (A) Oncoplot of recurrent somatic short-variant mutations detected in 4 MEITL tumors. Accompanying compounds that can target the corresponding gene mutations are labeled to the left of the oncoplot. (B) Recurrent somatic copy number alteration across the 4 MEITL using WGS data: copy-gain (left) and copy-loss (right) events. The vertical green line denotes the q = 0.25 (adjusted P value) cutoff for significant copy number events. (C) 17q arm-wide shows that large-scale, copy-neutral, loss-of-heterozygosity alterations were present and affected the STAT5B loci in all our MEITLs. (D) The 100% stacked bar plots comparing the proportions of known COSMIC (Catalogue of Somatic Mutations in Cancer) mutational signatures within each MEITL tumor. The denoted mutational signature numbers are as described at https://cancer.sanger.ac.uk/cosmic/signatures. (E) Circos (http://mkweb.bcgsc.ca/circos) plots depicting the structural rearrangements as links for each WGS sample. Links are colored differently for each type of structural rearrangement: red for duplications, blue for deletions, green for inversions, and black for interchromosomal translocations.
Copy number analysis with GISTIC28 revealed significant (q < 0.25) copy losses of 7p14.1, 8p23.1, 9p13.1, 9p12, and 14q11.2 and copy gain of 7p35 in the 4 MEITL tumors (Figure 1B). We also identified previously reported frequent chromosomal copy loss of 16q12.1 and gains of 8q24 (MYC locus) and 9q31.3 in 2 of 4 samples.7,9-11 Furthermore, the copy loss at 7p14.1 that encapsulated the TCR-γ alternate reading frame protein (TARP) gene coincided with previous studies on MEITL.12 Loss of 14q11.2 was reported in another study that suggested that this loss could be a diagnostic marker of T-cell lineage.13 We hypothesized that the loss of 7p14.1 and 14q11.2, which was observed in our MEITL samples, could be a consequence of physiological rearrangement of the TCR loci via VDJ (variable, diversity, joining) recombination14 (supplemental Table 4).
Within the 4 WGS MEITL samples, all harbored the STAT5B:p.N642H mutation. By imputing the single-nucleotide polymorphisms near STAT5B:p.N642, we found that the STAT5B loci were all copy-neutral loss of heterozygosity (Figure 1C). This phenomenon has been observed in JAK2 (9p24) and JAK3 (19p13) in other hematological neoplasms and is thought to arise from uniparental disomy events.15,16 This finding highlights the pivotal role of STAT5B in the development of MEITL.
Mutational signature analysis with Mutalisk17 was used to generate the mutational spectra of the samples (Figure 1D; supplemental Table 5). The age-related mutational signature18 was found to be dominant in our MEITL tumors, which coincided with the occurrence of MEITL in elderly individuals (median age, ∼60 years19 ), suggesting that MEITL tumors could have been “aging” in our patients over a long time. During this aging period, the tumors could have acquired heterogeneity, as observed in our previous study.7
Somatic structural rearrangement analysis found an average of 44 (range, 21-81) per sample. Genomic events of chromothripsis and chromoplexy were observed in MEITL13 and MEITL65, respectively. We also found a small cluster of structural breakpoints in the location of MYC in MEITL66 (Figure 1E; supplemental Figure 1). In particular, 2 interchromosomal translocations, from chromosomes 11 and 15, were found to fuse upstream of the MYC proto-oncogene. RNA-sequencing data also showed that MYC was significantly overexpressed (by 10.3 times) when compared with other MEITLs (supplemental Figure 2; supplemental Table 6).
Characterization of MEITL PDX model
We created a MEITL PDX model and an orthotopic model from case MEITL65 (Figure 2A). The PDX tumors retained the cytological atypia seen in the patient’s tumor throughout the series of passages, including round nuclei with dark, condensed chromatin; a scant-to-moderate amount of pale cytoplasm; and inconspicuous nucleoli (Figure 2A). The immunophenotype of the PDXs also remained concordant with the diagnosis: CD2+, CD3+, CD7+, CD8αα+, CD8β+, TIA1+, granzyme B+, TCRβ+, TCRγ+, CD56+, and MATK+, but CD4−, CD5−, CD79a−, and EBER− (supplemental Table 7; supplemental Figures 3 and 4). The proliferation fraction by Ki67 labeling hovered from 50% to 60%, similar to that of the patient’s tumor.
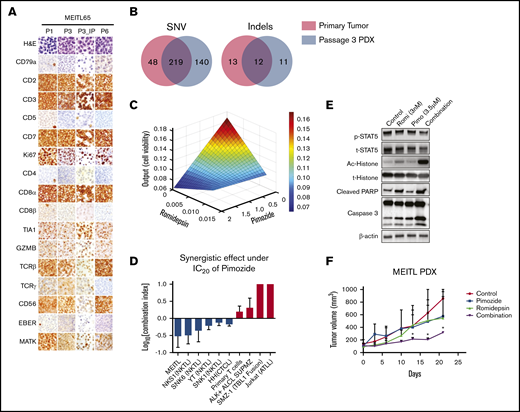
The synergistic effect of pimozide and romidepsin on a well-characterized MEITL PDX model. (A) Hematoxylin and eosin and immunohistochemical staining of subcutaneous MEITL PDX (P1, P3, and P6) and orthotopic (P3_IP) PDX tumors. (B) Venn diagrams of overlapping single-nucleotide variations (SNV, left) and indels (right) in a primary tumor (red) and a passage 3 subcutaneous tumor (blue). (C) Parabolic response surface maps of pimozide and romidepsin. (D) The combination index of romidepsin with the 20% inhibitory concentration (IC20) of pimozide. ALCL, anaplastic large-cell lymphoma; ATL, acute T-cell leukemia. Data are means ± standard deviation of 3 independent biological replicates. (E) Immunoblots of indicated proteins in MEITL tumor cells treated with control (dimethyl sulfoxide), pimozide, romidepsin, and combination of the 2 drugs for 48 hours at the indicated concentrations. The experiments were repeated at least 3 times. (F) The MIETL PDX tumor growth curves. Treatment with 25 mg/kg pimozide combined with 1 mg/kg romidepsin significantly decreased tumor growth compared with pimozide or romidepsin alone or the vehicle control (n = 3). Error bars indicate standard error of the mean. *P < .05.
The synergistic effect of pimozide and romidepsin on a well-characterized MEITL PDX model. (A) Hematoxylin and eosin and immunohistochemical staining of subcutaneous MEITL PDX (P1, P3, and P6) and orthotopic (P3_IP) PDX tumors. (B) Venn diagrams of overlapping single-nucleotide variations (SNV, left) and indels (right) in a primary tumor (red) and a passage 3 subcutaneous tumor (blue). (C) Parabolic response surface maps of pimozide and romidepsin. (D) The combination index of romidepsin with the 20% inhibitory concentration (IC20) of pimozide. ALCL, anaplastic large-cell lymphoma; ATL, acute T-cell leukemia. Data are means ± standard deviation of 3 independent biological replicates. (E) Immunoblots of indicated proteins in MEITL tumor cells treated with control (dimethyl sulfoxide), pimozide, romidepsin, and combination of the 2 drugs for 48 hours at the indicated concentrations. The experiments were repeated at least 3 times. (F) The MIETL PDX tumor growth curves. Treatment with 25 mg/kg pimozide combined with 1 mg/kg romidepsin significantly decreased tumor growth compared with pimozide or romidepsin alone or the vehicle control (n = 3). Error bars indicate standard error of the mean. *P < .05.
We used WES to further compare the genetic spectrum between the primary and PDX tumors. After removing the mouse-unique sequences with Xenome (https://hpc.nih.gov/apps/xenome.html), 219 short variants and 12 indels were common between the 2 tumors (Figure 2B; supplemental Table 8). We further validated 63 nonsilent short variants with Sanger sequencing (supplemental Table 9). Importantly, known driver mutations, including STAT5B, JAK3, SETD2, DUSP14, and CREBBP, were preserved in the PDX tumor. Altogether, the current results demonstrate that our MEITL PDX model preserved the principal histological and genetic characteristics of its original tumor and was genomically stable across passages, indicating its usefulness as an in vivo model for drug testing.
Evaluation of the therapeutic value of MEITL driver mutations
To evaluate the therapeutic potential of MEITL driver mutations, which were all preserved in our PDX model, we next performed a 12-drug, 3-level (dosages) QPOP, using targeted inhibitors together with chemotherapy drugs20-22 : pimozide,23 AZD1775,24 trametinib,7 tofacitinib,25 romidepsin,26 l-asparaginase, etoposide, panobinostat, cisplatin, gemcitabine, dexamethasone, and bortezomib. By correlating postdrug treatment cell viability values in a second-order quadratic equation, QPOP generates a ranked list of all possible drug combinations with their corresponding cell viability output values. The top 10 2-drug combinations are shown in supplemental Table S10. Pimozide and romidepsin combinations appear 3 times in the top 10 in the table (ranks 1, 5, and 6), suggesting that this combination is highly effective against the MEITL cells. This notion is supported by the response surface map of pimozide and romidepsin, which indicates synergistic drug interaction (Figure 2C). This synergistic effect was also observed in cutaneous T-cell lymphoma (CTCL) and natural killer T-cell lymphoma (NKTL), but not in primary T cells, Jurkat cells, and fusion gene–driven lymphomas. (Figure 2D; supplemental Figure 5). Western blot analysis showed that the cell apoptosis proteins cleaved caspase 3 and PARP were significantly upregulated in the combined treatment, but not in either single treatment (Figure 2E). Consistently, combination treatment of romidepsin and pimozide resulted in significant delay of tumor growth in our PDX model, which was not observed in the control or single-treatment groups (Figure 2F). Romidepsin was approved by the US Food and Drug Administration for peripheral T-cell lymphoma treatment in 2011. However, the response rate ranges from 41% to 58%, with considerable undesirable side effects.26,27 These data suggest that at a reduced dose, in combination with the STAT-5 inhibitor pimozide, could improve efficacy and reduce the side effects of the treatment of MEITL and peripheral T-cell lymphoma.
In summary, this study, to our knowledge, is the first WGS analysis of MEITL. We have established a comprehensive characterized MEITL PDX model, evaluated the targetable driver mutations in MEITL, and discovered a significant synergistic effect between romidepsin and pimozide, which may be applicable to NKTL and CTCL.
The WGS, WTS, and WES data have been deposited in the European Genome-Phenome Archive (accession numbers EGAS00001003876 [study] and EGAD00001005341 [data set]). The authors confirm that all data supporting the findings of this study are available in the article and the supplemental Material.
Acknowledgments
The authors thank all the participants in the study and Jeslin Chian Hung Ha, Rebecca Kee, and Khoo Lay Poh (Division of Medical Oncology, National Cancer Centre Singapore) for assistance in collating samples and clinical data from the patients.
This study was supported by research grants from the Singapore Ministry of Health, National Medical Research Council (NMRC-OFIRG16NOV090 [C.K.O. and S.T.L.], TCR/010-NCC/2012 [C.K.O, S.T.L., and S.Y.T.], and NMRC-OFLCG-18May0028 [C.K.O, S.T.L., S.Y.T., and E.K.-H.C.]), the TANOTO Foundation (NRDUKST18101) (C.K.O. and S.T.L.), the LING Foundation (NRDUKSN18101) (C.K.O. and S.T.L.), and the New Century Foundation (NCCRF-YR2014-SEP-SD2) (C.K.O. and S.T.L.).
Authorship
Contribution: D.H., J.Q.L., S.Y.T., and B.K.H.C. analyzed the data; D.H., J.Q.L., E.K.-H.C., S.Y.T., S.T.L., and C.K.O. conceived and designed the study; D.H., J.Q.L., K.H.B.M.K., Y.L., J.W.L.P., D.M.Z.C., E.K.Y.W., X.Z., J.G., S.Y.T., and J.Y.C. acquired the data; D.H., J.Q.L., J.G., S.Y.T., D.M.Z.C., and C.K.O. wrote the manuscript; and all authors approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Choon Kiat Ong, Division of Medical Oncology, National Cancer Centre Singapore, 11 Hospital Dr, Singapore 169610; e-mail: cmrock@nccs.com.sg; Soon Thye Lim, Division of Medical Oncology, National Cancer Centre Singapore, 11 Hospital Dr, Singapore 169610; e-mail: lim.soon.thye@singhealth.com.sg; Soo Yong Tan, Department of Pathology, National University Health System, 1E Kent Ridge Rd, Singapore 119228; e-mail: pattsy@nus.edu.sg; and Edward Kai-Hua Chow, National University Cancer Institute of Singapore, National University Health System, 5 Lower Kent Ridge, Singapore 119074; e-mail: csikce@nus.edu.sg.

