

Abstract
Machine learning (ML) is rapidly emerging in several fields of cancer research. ML algorithms can deal with vast amounts of medical data and provide a better understanding of malignant disease. Its ability to process information from different diagnostic modalities and functions to predict prognosis and suggest therapeutic strategies indicates that ML is a promising tool for the future management of hematologic malignancies; acute myeloid leukemia (AML) is a model disease of various recent studies. An integration of these ML techniques into various applications in AML management can assure fast and accurate diagnosis as well as precise risk stratification and optimal therapy. Nevertheless, these techniques come with various pitfalls and need a strict regulatory framework to ensure safe use of ML. This comprehensive review highlights and discusses recent advances in ML techniques in the management of AML as a model disease of hematologic neoplasms, enabling researchers and clinicians alike to critically evaluate this upcoming, potentially practice-changing technology.
Introduction
Despite recent research efforts, acute myeloid leukemia (AML) still poses a challenge in diagnosis and treatment alike, with curative options limited to a minority of cases.1 In the past, numerous preclinical and clinical studies, often with multicenter cohorts of patients, have led to a better understanding of AML pathogenesis and classification and, subsequently, to improved treatment options. The rise of genomics has further improved our understanding of AML and resulted in novel modes of risk stratification2 that were adopted in the European LeukemiaNet classification of AML.1
The first studies of ML techniques in the diagnosis of hematologic malignancies were conducted 2 decades ago. They started with the recognition of leukemic cells from blood samples,3,4 flow cytometry,5,6 and the evaluation of genetic data,7,8 establishing the groundwork of ML methods in the investigation of hematologic malignancies. However, computational power was limited, and an integration of different diagnostic modalities on multidimensional data sets seemed out of immediate reach. From the first theoretical introduction of an artificial neuron by McCulloch and Pitts in 1943,9 the refinement of computational methods and ML approaches in the last decades, especially in neural networks, has opened up a variety of integrative approaches in the field of hematology. The ever-growing body of data from clinical studies, as well as new insights from preclinical models, poses a challenge for researchers and clinicians alike to organize and interpret said data to improve patient care.
It has been shown that ML is well suited for dealing with large amounts of complex data and may prove to be a powerful tool in understanding and overcoming disease.10-12 Classically, diagnostic tests and patient data are interpreted by experienced clinicians who rely on years of medical education and training. However, ML algorithms have recently been shown to be on par with experts in a variety of tasks, from initial diagnosis, to prognosis estimation and prediction of treatment complications, to relapse monitoring in hematologic malignancies. However, many ML approaches have still not found their way into everyday clinical practice due to a variety of hurdles and pitfalls.
The current comprehensive review provides an overview of recent studies of ML in AML diagnostics, prognostication, and treatment allocation. It discusses current challenges and pitfalls to improve studies of ML in AML and foster safe and informed clinical use of the presented techniques in the future.
Diagnosis
Currently, the initial diagnosis of AML relies on 4 pillars: cytomorphology, cytogenetics, molecular genetics, and immunophenotyping.1 Given the evolving treatment stratification based on cytogenetic and molecular results, assigning patients to the best available treatment option seems appropriate.13 Therefore, precise characterization and classification of AML with high levels of accuracy are crucial for adequate therapy. Although our understanding of cancer in general has improved with the ever-increasing amount of genetic and genomic data, we still struggle to implement these large and complex data sets into clinical practice. ML approaches have shown tremendous potential in the analysis of complex genetic data.14
Analyzing >12 000 samples from >100 different studies, Warnat-Herresthal et al15 combined transcriptomic and genomic data with ML to develop classifiers that accurately detect AML in a near-automated and low-cost method. However, not every center interested in research in the applications of ML in cancer has such large data sets at hand. Fortunately, various freely accessible online data sets are available for multiple research purposes; these include the Leukemia Gene Atlas,16 Beat-AML,17 and The Cancer Genome Atlas.18 They allow researchers all across the globe to evaluate genetic risk profiles or identify novel genetic targets for individualized cancer therapy with the aid of ML techniques.19 This option could prove useful in the development of prospective basket trials of specific cancer-type overlapping mutations identified by ML algorithms. Support vector machines (SVMs), an ML technique that delineates data points in a coordinate system by calculating a hyperplane between distinct data sets, can be used for classification of high-dimensional data sets.20 They can be applied to classify subtypes in large genomic data sets once pre-processing steps such as filtering for biomarker signatures or gene alterations have been performed to organize multidimensional data sets; the SVMs use these for classification, thereby revealing potential targets for therapy,21,22 and detect leukemic stem cells by genetic profiling.23
A well-known disease-causing mutation of the FMS-like tyrosine kinase 3 (FLT3) occurs in almost one-third of AML cases, with internal tandem duplication (ITD) representing the most common FLT3 mutation.24 The combination of RNA-sequencing and genotyping with ML can distinguish malignant cell types and identify prototypic genetic lesions and an association of FLT3-ITD with progenitor-like cells.25 SVM and random forest (RF), a combination of decision trees in which each tree depends on the values of a random vector sampled independently and with the same distribution for all trees in the forest,26 are able to identify feature genes with the capacity to predict the mutation status of FLT3-ITD.27
Deep neural networks (DNNs) can identify critical proteins associated with FLT3-ITD.28 DNNs are a subset of ML that imitate the neuronal structure of the brain by creating interconnected artificial neural networks that can be applied for computer vision purposes, especially object detection, image segmentation, and classification.29 After adequate pre-processing of image data, DNNs can be used in computer-aided diagnosis in cytomorphology. Key steps in DNN-based assessment of bone marrow and peripheral blood smears are cell segmentation, extraction, quantification of cell-specific features, and subsequent cell classification.30 Especially in leukemia, precise recognition of white blood cells with various segmentation techniques (filtering, enhancement, edge detection, feature extraction, and classification)31 is crucial for correctly distinguishing between leukemic and non-leukemic cells.32-34 ML can use these techniques to analyze whole slides with automated focusing.35 Classification of leukemia subtypes (AML, acute lymphoblastic leukemia, chronic myeloid leukemia, and chronic lymphocytic leukemia) can be achieved by a variety of ML approaches such as DNN,36 SVM, and k-means-clustering (an unsupervised ML technique in which similar data points are grouped into k clusters according to their distance to a cluster mean).37,38
Another essential part of the diagnostic process in AML is flow cytometry,39 which can aid in the detection of relapse with a higher sensitivity than cytomorphology alone.40 ML can be used to precisely distinguish between samples from AML patients and healthy individuals.41-43 Computer-driven analysis of flow cytometry using clustering techniques (eg, FlowSOM) in combination with ML techniques (eg, SVM and RF) increases diagnostic accuracy in various hematologic malignancies44 and correctly classifies rare cells.45 FlowSOM is based on self-organizing maps to analyze flow or mass cytometry data, providing an overview of large sets of markers,46 and it thereby aids in phenotyping leukemia and assessing measurable residual disease (MRD).47 ML may thus provide an automated classification of data generated by flow cytometry and aid clinicians in their analysis and interpretation of the data by providing them with various differential diagnoses and their respective likelihood based on the given data. Hence, an integration of all diagnostic modalities in the evaluation of AML by the combination of different ML techniques seems feasible and provides a fast, automated, data-driven overview of each individual suspected case of AML for the medical professional to evaluate and verify.
Treatment and prognosis
The European Leukemia Net 2017 risk stratification divides patients with AML into favorable, intermediate, and adverse risk groups with distinct therapeutic implications and outcomes.1 ML is advantageous in the early detection of potentially high-risk leukemias based on their individual genetic profile. Morita et al48 analyzed bone marrow samples of 868 patients with myeloid leukemias (AML, myelodysplastic syndrome [MDS], chronic myelomonocytic leukemia, and myeloproliferative neoplasm) and generated an ML-based model that accurately predicts clinical phenotype based on somatic mutation data. Siddiqui et al49 proposed an ML model based on clinical parameters known before treatment that predicts mortality rates for patients undergoing chemotherapy, thereby enabling clinicians to identify patients suitable for intensive induction regimens. DNN approaches have been shown to accurately predict AML prognosis based on cytogenetics, mutational status, and age.50 Gerstung et al51 have reported that large data sets combining clinical and genomic data in the form of knowledge banks can therefore be used to guide clinicians to precisely tailor a treatment approach for the individual patient; this method provides an accurate prediction of relapse, remission, and overall survival.
ML may even yield higher levels of accuracy compared with current standards. Fleming et al52 used RF and decision trees to predict survival prognosis in >2000 cases of non-APL/AML and reported a lower error rate for their model compared with the European Leukemia Net 2017 score. Similarly, Shreve et al53 devised an ML model based on clinical, cytogenetic, and mutational data to predict personalized outcomes for the individual patient and reported a significantly better performance than the European Leukemia Net classification. Li et al54 developed an algorithm for automatic classification of AML, MDS, and healthy samples based on >2000 patients limiting the number of flow cytometry markers while maintaining high levels of accuracy.
ML can be used to develop novel prognostic indices or refine the understanding of already established prognostic mutational markers. NPM1 mutations are among the most commonly found mutations in AML, representing a distinct entity in the World Health Organization classification.55 Patkar et al56 identified genomic aberrations in NPM1mut AML and developed a scoring system classifying NPM1mut AML into 3 prognostic subgroups. Wagner et al57 used an ML approach to associate a 3-gene expression signature consisting of CALCRL, CD109, and LSP1 with overall survival, resulting in a prognostic score that includes gene expression levels and clinical data.
However, AML therapy remains challenging, and refractory disease poses a substantial threat for patient outcome.58 ML can predict the likelihood of complete response in pediatric AML patients who received induction therapy based on gene expression patterns obtained through RNA sequencing.59 Based on proteomics, ML can divide patients with AML into different treatment response groups, although combined use with clinical data may be essential.60 MRD is an important marker for risk stratification and decision-making concerning therapeutic adjustments.61,62 Measurement of MRD can be improved by ML techniques such as SVM,63-65 and the evaluation of MRD is of growing importance in clinical decision-making in the management of AML. However, MRD evaluation is not available at all sites because it requires a high level of accuracy and technical expertise that can thus far only be achieved by specially equipped laboratories. ML techniques help implement MRD assessment in clinical practice by providing a highly standardized and data-driven approach based on the evaluation of large multicenter MRD data sets. Therefore, collaborative prospective studies between experienced laboratories and hematologic centers are needed to establish ML models that can accurately assess MRD; the goal is to provide to the clinical practice high-quality, standardized and automated MRD assessment verified by field experts.
Recently, a variety of novel therapeutic agents have been approved by both the US Food and Drug Administration and the European Medicines Agency for frontline treatment of patients with AML; long-term benefits remain uncertain, however, and study design as well as eligibility criteria may be flawed.66-68 ML provides the means to improve patient selection, recruitment, and monitoring in clinical trials by assessing eligibility criteria, scanning electronic health records for suitable patients, or predicting the likelihood of failure or success in a trial.69 ML models have been established in drug discovery and development, and DNNs especially show tremendous potential in identifying biomarkers and druggable targets and in the assessment of potential therapeutic molecules.70,71 The National Cancer Institute and the Dialogue on Reverse Engineering Assessment and Methods (DREAM) have launched challenges to develop ML tools to discover novel treatment strategies and detect drug-sensitive targets from genomic data.72 ML can use these large genomic data sets to predict targets for therapeutic agents. Lee et al73 identified SMARCA4 as a marker and driver of sensitivity to the topoisomerase II inhibitors mitoxantrone and etoposide, showing increased drug sensitivity both in ML models and in in vitro assays. Chen et al74 used ML to assess potential STAT3 inhibitors in AML and MDS. Janssen et al75 developed drug discovery maps based on t-distributed stochastic neighbor embedding to predict novel inhibitors of FLT3, and Cutler and Fridman76 generated an ML model that predicts high sensitivity to FLX925, a small molecule inhibitor of FLT3, in AML.
Despite the advent of targeted therapy, in the majority of AML cases, a curative treatment approach is still often limited to allogeneic stem cell transplantation, which harbors various risks. These risks include high treatment toxicity, infectious complications, graft-versus-host-disease (GVHD), transplant failure, and relapse.77
Evaluating suitable patients for transplantation and patients at risk for complications is therefore crucial before starting conditioning therapy. Shouval et al78 identified key variables to predict overall survival 100 days after transplantation in an analysis of >25 000 leukemia patients from the European Society for Blood and Marrow Transplantation with various ML techniques; they validated the scoring system in a prospective cohort study of 1848 patients from the Italian national transplant network.79 The choice of conditioning regimen and post-grafting immunosuppression may therefore be guided by ML algorithms to design a personally tailored approach for the individual patient based on large databases of specific immunogenetic environments of patients undergoing allogeneic hematopoietic stem cell transplantation.80 Relapse after transplantation can be estimated by using alternating decision trees.81 ML can also be used to predict development of acute GVHD after allogeneic transplantation82 and stratify outcomes in chronic GVHD, revealing novel groups at risk based on clinical phenotypes more accurately than current approaches based on cumulative severity.83
Discussion
ML has already proven to be a versatile, precise, and robust tool in the diagnostic and therapeutic evaluation of AML, with a variety of challenges for future research as summarized in Table 1.
Applications and challenges of ML in the management of AML
| Application | Cytomorphology/histology | Immunophenotyping | Clinical data | Cytogenetics/molecular genetics | New therapies/prognostic scores |
|---|---|---|---|---|---|
| Improvements needed | Precision of image segmentation (eg, detection of cell boundaries) | MRD evaluation (eg, standardization of cutoffs to form a decision boundary) | Integration of different high-dimensional data sets | Availability of data | Prospective studies for validation |
| Number values such as laboratory results as well as written text are better evaluated by different ML techniques (integrative models needed) | Models trained on online data (eg, Beat-AML or The Cancer Genome Atlas) may not be accurate on regional data | Majority of ML studies are only retrospective; models have to be evaluated in a prospective manner to evaluate their translational application in patient care | |||
| Feature extraction (eg, relation of nucleus to cytoplasm) | Classification methods | Standardization of clinical reports | Pre-processing of high-dimensional data | ||
| Which combination of different ML techniques shows the most accurate results? | Uniformity of clinical reports (eg, with standardized vocabulary) makes natural language processing easier | Accurate filtering of biosignatures is needed before classification | |||
| Cell classification | |||||
| Labeling by field experts needed | |||||
| Outlook | • Integrated workflow of various ML techniques to guide clinical decision-making | ||||
| • Strict legal and regulatory framework to ensure patient safety | |||||
| • Prospective clinical trials to verify robustness of ML models | |||||
| • Physicians with basic knowledge in ML techniques to optimally implement ML into clinical practice | |||||
| Application | Cytomorphology/histology | Immunophenotyping | Clinical data | Cytogenetics/molecular genetics | New therapies/prognostic scores |
|---|---|---|---|---|---|
| Improvements needed | Precision of image segmentation (eg, detection of cell boundaries) | MRD evaluation (eg, standardization of cutoffs to form a decision boundary) | Integration of different high-dimensional data sets | Availability of data | Prospective studies for validation |
| Number values such as laboratory results as well as written text are better evaluated by different ML techniques (integrative models needed) | Models trained on online data (eg, Beat-AML or The Cancer Genome Atlas) may not be accurate on regional data | Majority of ML studies are only retrospective; models have to be evaluated in a prospective manner to evaluate their translational application in patient care | |||
| Feature extraction (eg, relation of nucleus to cytoplasm) | Classification methods | Standardization of clinical reports | Pre-processing of high-dimensional data | ||
| Which combination of different ML techniques shows the most accurate results? | Uniformity of clinical reports (eg, with standardized vocabulary) makes natural language processing easier | Accurate filtering of biosignatures is needed before classification | |||
| Cell classification | |||||
| Labeling by field experts needed | |||||
| Outlook | • Integrated workflow of various ML techniques to guide clinical decision-making | ||||
| • Strict legal and regulatory framework to ensure patient safety | |||||
| • Prospective clinical trials to verify robustness of ML models | |||||
| • Physicians with basic knowledge in ML techniques to optimally implement ML into clinical practice | |||||
The efficiency of ML algorithms greatly depends on the quality and quantity of data they are trained on, as well as the selected end points and outcomes that researchers use. Therefore, large data sets are needed to construct and train such models.84 With publicly available data sets, researchers have access to large amounts of training data for the development of ML tools, providing even small centers with the opportunity to conduct research in ML. Nevertheless, it is questionable how well ML models developed on online data sets can perform in a regional setting. ML algorithms can automatize narrow repetitive tasks and thereby aid clinicians in accurately diagnosing AML as well as cutting time and effort in diagnostic steps such as the assessment of genetic, mutational, cytomorphologic, and flow cytometry data. The combination of different diagnostic modalities is crucial for the correct diagnosis of AML. An implementation of different ML techniques linking, for example, the results of flow cytometry, cytomorphology, and cytogenetics can provide clinicians with integrated ML tools to evaluate each suspected case of AML faster and with higher precision because adaptive ML tools are able to learn with every new case they are trained on, thus improving accuracy.
Furthermore, ML algorithms will help to better understand the complex interaction of distinct molecular subgroups in AML by identifying specific markers delineating specific groups of patients. These markers can also be evaluated in different hematologic entities (eg, MDS), and discoveries of overlapping disease-driving genetic alterations provide the opportunity for the development of prospective basket trials to create a directed therapy against disease-causing genes. Prognostic indices based on patient features derived by ML promise an unbiased view of potential markers of risk and adverse outcome and may refine current standards. Clinicians benefit from such tools as they partially free them from handling large amounts of data and equip them with methods to match individual patients with an ideal treatment.12 ML therefore provides a powerful tool in the advent of precision medicine by identifying disease-specific genetic alterations and simultaneously recommending molecular structures that may be used to target these mutations for the individual patient. Once established, these algorithms could even provide hematologic expertise to regions without immediate access to large medical centers and help general practitioners to adequately screen for patients in need of hematologic assessment or treatment.
Nevertheless, tight regulation and oversight are crucial for the proper application of computer-aided diagnosis and treatment allocation. A sophisticated structure of regulatory oversight, legal frameworks, and monitoring systems, adaptive to the fast pace of the current developments, is of the highest importance to ensure safe development and use of ML in everyday hematologic practice. The fast development of ML not only in the field of hematology but overall medical practice shows that computer skills are essential in medical training. We argue that basic knowledge of ML techniques, especially their potential in diagnostics and therapeutics as well as their limitations therein and potential for bias, is a key skill in medical education preparing for current and future developments and changes in practice. Future physicians should be taught to be critical users of ML in their practice, understanding how different models work, and what is in and out of reach of ML; this would enable them to properly integrate these methods into their practice and critically analyze and evaluate the data and recommendations provided by these tools.
As promising as these first results of ML in hematology may be, however, there is still a long road ahead. Clinicians and researchers should be aware of common pitfalls in ML when designing new studies.85 For instance, many techniques require splitting research data into a training set and a testing set, and sometimes inadequate splits or hidden trends in data sets can falsify results. Researchers should also be wary for seemingly insignificant hidden variables that can influence ML models (eg, the placing of the scale bar in microscopic images). Furthermore, ML models can be overfit by feeding them biased data or making them catch “noise” instead of actual features, which results in a model that does suspiciously well in training sets but often cannot generalize well in test sets.86 There is no gold standard in model selection, and every ML set-up depends on the use-case and research question at hand. It is also important to report on negative results and difficulties, given the variability of different ML set-ups, to work out collectively which approaches are more promising in which specific use-case. Many studies of ML in AML and cancer in general are still only retrospective and the underlying code of the ML model is often not reported, and therefore reports are at risk of bias.87 Prospective validation in a proof-of-concept fashion is needed. ML-derived scoring systems, as well as individually tailored treatment approaches, can be verified in prospective trials proving or disproving the robustness of different models. Stand-alone ML tools based only on retrospective data are insufficient for widespread clinical use.
From a software point of view, prospective validation poses a challenge, because the large variety of available ML technology offers not only a vast amount of models to choose from but also may lead to flaws in study design because the optimal model for a distinct research question might not be chosen. At first, when conducting ML-based research, it is usually unknown which technology and parameterization lead to sufficient quality, especially for smaller data sets.88 Therefore, software solutions should provide an iterative workflow to improve in a step-wise manner the ML set-up and integrate a broad spectrum of technology in a uniform way to support technological variety and progress.89,90
As shown in Figure 1, an ML workflow consists of several data preprocessing and postprocessing steps, as well as meta-mechanics to optimize parameters and track objectives.91,92 To increase the transparency of the approach and results, and to ensure reproducibility as well as comparability, a more abstract technical workflow description is required (eg, based on attribute grammars or model-based development approaches). Finally, domain experts (ie, physicians) need to have direct access to the ML workflow, as the translation of medical requirements, knowledge, and objectives by technicians implies obstacles and sources of errors. Hence, this should be minimized by adaptive, context-sensitive, and customizable user interfaces. Cooperation between study groups and a pooling of data sets may yield even more robust results.
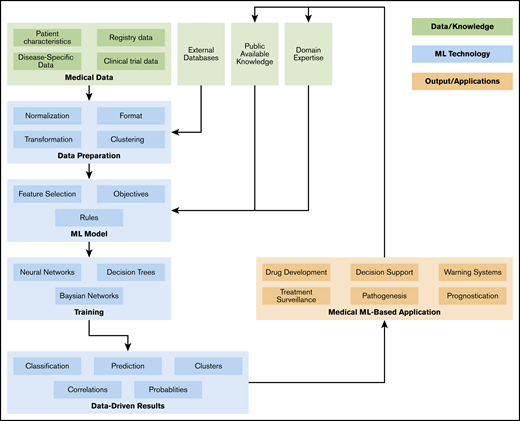
In conclusion, ML in AML introduces a variety of novel and deeper insights in disease development and has the potential to significantly improve prognostication, personalized treatment, and patient surveillance. Close cooperation between computer scientists, data scientists, software developers, basic medical researchers, and physicians is imperative for sustained success and regulatory oversight. Legal frameworks are needed for safe and standardized development and use of ML tools for medical practice. The awareness of potential pitfalls of ML techniques and the knowledge gained from recent studies should lead to a more informed design of ML research. The goal is to create integrative tools that can analyze and interpret data from multidimensional diagnostic modalities to further aid the clinician in everyday practice in diagnosis, prognostication, and treatment allocation of patients with AML, ultimately improving patient outcome.
Requests for data sharing may be submitted to the corresponding author (Jan-Niklas Eckardt; e-mail: jan-niklas.eckardt@uniklinikum-dresden.de).
Authorship
Contribution: J.-N.E. performed the literature search and wrote the draft; J.M.M., M.B., and K.W. edited the manuscript; and all authors revised and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jan-Niklas Eckardt, Department of Internal Medicine I, University Hospital Carl Gustav Carus, Fetscherstr 74, 01307 Dresden, Germany; e-mail: jan-niklas.eckardt@uniklinikum-dresden.de.

