

Key Points
A combination cell-viability screen for copanlisib partners identified venetoclax as the strongest one in BCLs.
These data support the rationale for the SAKK 66/18 phase 1 study in patients with relapsed/refractory lymphomas.

Abstract
Copanlisib is a pan–class I phosphoinositide 3-kinase (PI3K) inhibitor with preferred activity toward PI3Kα and PI3Kδ. Despite the clear overall clinical benefit, the number of patients achieving complete remissions with the single agent is relatively low, a problem shared by the vast majority of targeted agents. Here, we searched for novel copanlisib-based combinations. Copanlisib was tested as a single agent, in combination with an additional 17 drugs in 26 cell lines derived from mantle cell lymphoma (MCL), marginal zone lymphoma (MZL), and T-cell lymphomas. In vivo experiments, transcriptome analyses, and immunoblotting experiments were also performed. Copanlisib as a single agent showed in vitro dose-dependent antitumor activity in the vast majority of the models. Combination screening identified several compounds that synergized with copanlisib. The strongest combination was with the B-cell lymphoma 2 (BCL2) inhibitor venetoclax. The benefit of the combination over single agents was also validated in an MZL xenograft model and in MCL primary cells, and was due to increased induction of apoptosis, an effect likely sustained by the reduction of the antiapoptotic proteins myeloid cell leukemia 1 (MCL1) and BCL-XL, observed in MCL and MZL cell lines, respectively. These data supported the rationale for the design of the Swiss Group for Clinical Cancer Research (SAKK) 66/18 phase 1 study currently exploring the combination of copanlisib and venetoclax in relapsed/refractory lymphomas.
Introduction
The phosphatidylinositol 3-kinases (PI3Ks) are composed of a catalytic subunit complexed with a regulatory subunit that regulates the activity, localization, and binding of the dimer.1 There are 4 different class I isoforms (p110α, p110β, p110δ, p110γ) of the catalytic subunit, which represent therapeutic targets to pharmacologically block PI3K signaling.1 In lymphomas, the PI3K pathway is crucial in the signaling cascade downstream not only to the B-cell receptor but also to other receptors such as cytokine receptors.1,2 PI3Kδ is mostly expressed in B cells, and the PI3Kδ inhibitor idelalisib (CAL-101, GS-1101) was the first PI3K inhibitor approved by the US Food and Drug Administration (FDA) for patients with relapsed follicular lymphoma (FL) with 2 or more prior therapies based on an overall response rate (ORR) of 57% with 7% of complete remission (CR).3,4 Similar results were seen in patients with relapsed marginal zone lymphoma (MZL) (ORR, 47%; no CR)4 and in relapsed/refractory mantle cell lymphoma (MCL) (ORR, 40%; CR, 5%).5 PI3Kδ selectivity represents a limit for the antilymphoma activity of idelalisib, as shown by the high expression of other catalytic subunits in resistant cases.6-8 Compounds targeting >1 isoform present a broader pattern of preclinical antitumor activity in B-7-11 and T-cell malignancies.8,12,13 Copanlisib (BAY 80-6946) is a pan–class I PI3K IV inhibitor with dominant activity toward PI3Kα and PI3Kδ.14,15 Copanlisib has also shown preclinical antitumor activity in diffuse large B-cell lymphoma (DLBCL)7,10 and chronic lymphocytic leukemia (CLL).11 The early demonstration of clinical activity in FL and DLBCL16 has been confirmed in phase 2 studies and extended to MZL, MCL, small lymphocytic lymphoma, and peripheral T-cell lymphoma (PTCL).17-19 The toxicity of copanlisib (hyperglycemia, diarrhea, hypertension, and neutropenia as the most commonly observed side effects) compares well vs other agents of the same class and it has fewer and less severe gastrointestinal toxicities than idelalisib.19-22 Copanlisib is now FDA approved for relapsed FL patients after at least 2 systemic therapies due to the ORR of 59% with 14% of CR achieved in the phase 2 study.18
The usual low CR rate achieved with small molecules given as single agents16-18,23 is consistent with the notion that targeting a single pathway is unlikely to eradicate tumor cells due the activation of additional pathways.1,24 With the aim of identifying combinations that can increase the cure rate, we performed a small-molecule combination screen in non-DLBCL lymphoma models that identified synergistic copanlisib combinations and provided the rationale for the Swiss Group for Clinical Cancer Research (SAKK) 66/18 phase 1 study currently exploring the combination of copanlisib and venetoclax in relapsed/refractory lymphomas (NCT03886649).
Material and methods
Cell lines
Cell lines derived from MCL (JEKO1, Rec1, JVM2, Granta519, Maver1, Mino1, SP-49, SP-53, UPN1, Z138), MZL (Karpas1718, VL51, SSK41, ESKOL, HAIR-M, HC-1), CLL (MEC1), anaplastic large cell lymphoma (ALCL) (SU-DHL-1, L82, MAC1, KARPAS299, KI-JK), PTCL (FEPD, HH) and Sezary syndrome (SS) (H9, HUT78) were used. All media were supplemented with fetal bovine serum (10%), penicillin-streptomycin-neomycin (∼5000 U of penicillin, 5 mg of streptomycin, and 10 mg of neomycin per milliliter; Sigma-Aldrich, Buchs, Switzerland), and l-glutamine (1%). Cell identity was authenticated by short tandem repeat DNA profiling (IDEXX BioResearch, Ludwigsburg, Germany).
Compounds
Venetoclax (ABT199), bortezomib, crizotinib, ibrutinib, MI2, palbociclib, panobinostat, romidepsin, ruxolitinib, AZD1208, and MIK665 were obtained from Selleckchem (Houston, TX). Bendamustine was obtained from Sigma-Aldrich. Lenalidomide was obtained from Santa Cruz Biotechnologies. Copanlisib, roniciclib (BAY 1000394), bromodomain and extraterminal motif (BET) inhibitor BAY 1238097, positive transcription elongation factor b (PTEFb) inhibitor atuveciclib (BAY 1143572), and AKT inhibitor (BAY 1125976) were provided by Bayer AG.
In vitro antitumor activity
Cell lines were plated in 384-well plates in appropriate growth medium. Test compounds were added to cells by means of an HP D300 digital dispenser in a 10-step 2.5-fold dilution series. Cells were treated for 72 hours followed by assessment of cell viability by means of Cell Titer Glo assay (Promega, Madison, WI). Experiments were conducted in duplicates. Synergy was assessed with the Chou-Talalay combination index: synergism (<0.9), additive (0.9-1.1), antagonism/no benefit (>1.1).25 For sequential experiments, cells were treated with copanlisib for 24 hours and washed by centrifuge (1500 rpm, 10 minutes) followed by removal of the supernatant and venetoclax treatment of an additional 24 hours, or with the inverse drug sequence. A parallel experiment was performed for 48 hours incubating the cells with both drugs simultaneously. Cell viability was measured as previously described.8
Primary cells
Peripheral blood was obtained from patients who gave written informed consent for their blood products to be used for research under an institutional review board–approved protocol. Primary cells were isolated by Ficoll density-gradient centrifugation and cultured for 48 hours; the percentage of viable and apoptotic cells (Annexin V+/7-aminoactinomycin D negative [7AAD−] and Annexin V+/7AAD+) was determined by double staining the cells with Annexin V–fluorescein isothiocyanate/7AAD.
In vivo experiments
Female NODscid mice (18-25 g, 6-8 weeks) from Janvier (Le Genest-Saint-Isle, France) were injected subcutaneously in the left inguinal region with 5 × 106 JEKO1 cells in 0.1 mL of 50% Matrigel/50% medium or 1.5 × 107 SSK41 cells in 0.1 mL of 100% Matrigel. Mice were randomized (n = 9-10 per group) at an average tumor area ∼50 mm2 and treated orally (per os) or IV for 14 days with vehicle or compounds according to treatment schedule. Animal experiments were approved by the relevant regulatory agency of the federal state of Berlin (Landesamt für Gesundheit und Soziales Berlin). Mice body weight was determined daily, and body weight loss ≥ 20%, compared with the first day of therapy, was judged excessive and the dose considered as toxic. Tumors were measured twice a week using a caliper and volume was calculated using the formula: tumor volume = [length × width × width]/2. Differences in tumor volumes were calculated using the Wilcoxon rank-sum test (Stata/SE 12.1 for Mac; Stata Corporation, College Station, TX). The P value for significance was <.05. Graphs were obtained with GraphPad Prism (v. 7.0d; GraphPad Software, La Jolla, CA).
Western blotting analysis
Western blots of cell-line protein extracts were performed as previously described,8 using antibodies against PI3K p110α, PI3K p110γ, AKT, phosphorylated Akt (pAkt; Ser473), and myeloid cell leukemia 1 (MCL1) (Cell Signaling Technology, Danvers, MA); PI3K p110δ, B-cell lymphoma 2 (BCL2; N-19), and BCLXL (H-5) (Santa Cruz Biotechnology, Santa Cruz, CA); poly (ADP-ribose) polymerase (PARP) (BD Biosciences, Allschwil, Switzerland); glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Ritomed, Berlin, Germany); PI3K p110β (Millipore, Darmstadt, Germany); and vinculin (Sigma-Aldrich).
Transcriptome profiling
RNA was extracted and processed for RNA sequencing (stranded, single-ended 75-bp long sequencing reads) using the NEBNext Ultra Directional RNA Library Prep kit for Illumina (New England BioLabs Inc, Ipswich, MA) on a NextSeq 500 (Illumina, San Diego, CA) as previously described.26 Data mining was performed as previously described.27 Significantly differentially expressed transcripts had absolute log fold change (FC) > 0.25 and Benjamini-Hochberg multiple test corrected P values (false discovery rate [FDR]) <.05. Functional annotation was done using gene set enrichment analysis on FC preranked lists with gene sets from the MSigDB 5.2 Hallmark collection28 and from different publications,8 applying as thresholds an absolute normalized enrichment score > 1.5 and P and FDR values < 0.05.
Real-time PCR
Real-time polymerase chain reaction (PCR) was performed as previously described29 using the QuantiFast SYBR Green PCR kit (Qiagen, Germantown, MD). Used primers were: 5′-CCTAGCCATAGAGCTTCCTTTC-3′ (PIK3IP1-forward), 5′-TTCTCTTCTCCAGCATCACTAAC-3′ (PIK3IP1-reverse), 5′-CGACCACTTTGTCAAGCTCA-3′ (GAPDH-forward), 5′-CCCTGTTGCTGTAGCCAAAT-3′ (GAPDH-reverse). Primers were purchased from Sigma-Aldrich.
Data mining
Associations in 2-way tables were tested for statistical significance using either the χ2 test or Fisher's exact test (2-tailed), as appropriate. Binomial exact 95% confidence intervals (CIs) were calculated for median percentages. Differences in 50% inhibitory concentration (IC50) values among subtypes were calculated using the Wilcoxon rank-sum test. Baseline gene-expression levels of PI3K isoforms were extracted from the GSE946698 data set obtained using the HumanHT-12 v4 Expression BeadChip (Illumina). Statistical significance was defined by P < .05. Statistical analyses were conducted using Stata/SE 12.1 for Mac (Stata Corporation, College Station, TX).
Results
Copanlisib has antineoplastic activity as a single agent in lymphomas in vitro
Following the demonstrated preclinical activity of copanlisib in DLBCL cell lines,7 we have explored its antineoplastic activity as a single agent in 26 additional lymphoma models, derived from B- and T-cell malignancies. The B-cell lymphomas comprised MCL (n = 10), MZL (n = 6), and CLL (n = 1). The T-cell lymphomas comprised anaplastic large cell lymphoma (ALCL; ALCL−ALK+ [n = 4]; ALCL−ALK− [n = 1]), PTCL (n = 1), and cutaneous T-cell lymphoma (CTCL; n = 3, including 2 SS cases). Copanlisib was active both in B-cell– and T-cell–derived lymphomas; however, the median IC50 in the B-cell lymphomas was lower than the median IC50 derived from the T-cell lymphomas (P = .0002) (Table 1; supplemental Table 1).
Antitumor activity of copanlisib in lymphoma cell lines other than DLBCL
| No. of cell lines | Median IC50, nM | 95% CI, nM | |
|---|---|---|---|
| All cell lines | 26 | 75 | 21-160 |
| B-cell lymphomas | 17 | 22 | 15-99 |
| MCL | 10* | 21.5 | 3-103 |
| MZL | 6† | 36 | 6-175 |
| CLL | 1‡ | 23 | n.d. |
| T-cell lymphomas | 9 | 285 | 137-926 |
| CTCL | 3§ | 304 | 131-970 |
| ALCL, ALK+ | 4|| | 215 | 50-285 |
| ALCL, ALK− | 1¶ | 399 | n.d. |
| PTCL-NOS | 1# | 1663 | n.d. |
| No. of cell lines | Median IC50, nM | 95% CI, nM | |
|---|---|---|---|
| All cell lines | 26 | 75 | 21-160 |
| B-cell lymphomas | 17 | 22 | 15-99 |
| MCL | 10* | 21.5 | 3-103 |
| MZL | 6† | 36 | 6-175 |
| CLL | 1‡ | 23 | n.d. |
| T-cell lymphomas | 9 | 285 | 137-926 |
| CTCL | 3§ | 304 | 131-970 |
| ALCL, ALK+ | 4|| | 215 | 50-285 |
| ALCL, ALK− | 1¶ | 399 | n.d. |
| PTCL-NOS | 1# | 1663 | n.d. |
n.d., not determined; PTCL-NOS, PTCL–not otherwise specified.
GRANTA-519, JEKO1, JVM-2, MAVER-1, MINO, REC-1, SP-49, SP-53, UPN-1, Z-138. †KARPAS1718, VL51, SSK41, ESKO-L, HAIR-M, HC-1. ‡MEC-1. §H9, HH, HUT78. ||Ki-JK, KARPAS-299, L-82, SU-DHL-1. ¶MAC-1. #FE-PD.
Looking at the PI3K isoforms at protein level (supplemental Figure 1), PI3Kα was more expressed in B- than in T-cell lymphomas (P = .049), whereas no differences were observed for the other isoforms. At RNA levels, PI3Kα (P = .042), and PI3Kδ (P = .042) were higher in B- than T-cell lymphomas, whereas the opposite was true for PI3Kβ (P < .001); PI3Kγ RNA levels did not show any difference. Sensitivity to copanlisib was positively correlated with PI3Kα protein expression (R = 0.47; P = .019) and negatively to PI3Kβ RNA levels (R = −0.62; P < .001) only when considering all the lymphomas together. However, no correlation was observed within the individual B- or T-cell groups, indicating that the differences were due to the different isoform expression.
Taking advantage of previously reported data,8 the activity of copanlisib was compared with PI3Kδ inhibitor idelalisib and to the dual PI3K/mammalian target of rapamycin (mTOR) inhibitors bimiralisib and apitolisib. A strong correlation in the pattern of activity across 19 cell lines was observed between copanlisib and idelalisib (R = 0.77, P < .001), copanlisib and bimiralisib (R = 0.64; P = .003), and copanlisib and apitolisib (R = 0.6; P = .006) (supplemental Figure 2). However, copanlisib was the most active compound in terms of IC50 values (P < .05) (supplemental Figure 2).
Across all cell lines, PI3Kβ RNA levels were negatively correlated with the sensitivity to both idelalisib (R = −0.53; P = .023) and bimiralisib (R = −0.48; P = .046), whereas the antitumor activity of the PI3Kδ inhibitor was positively correlated with PI3Kδ RNA levels (R = 0.61; P = .007). Correlations were not confirmed within the B- and T-cell groups. No significant correlations were seen for the sensitivity to apitolisib.
Copanlisib has in vivo antitumor activity as single agent
The observed in vitro activity was confirmed in vivo using the human JEKO1 MCL xenograft model. Animals treated with copanlisib (10 mg/kg, IV, 2 days on/5 days off) experienced a reduction in the tumor growth compared with the control group (IV vehicle, 2 days on/5 days off) starting at day 4 and persisting to the last time point (day 14) (P < .05) (supplemental Figure 3A). No significant body weight loss was observed (supplemental Figure 3B).
Copanlisib synergizes with venetoclax and other compounds
After determining single-agent activity of copanlisib, we evaluated the single-compound activity of a series of potential combination partners, including approved and experimental inhibitors of key regulatory pathways: the AKT1/2 inhibitor BAY 1125976, the BCL2 inhibitor venetoclax, the BET inhibitor BAY 1238097, the BTK inhibitor ibrutinib, the cyclin-dependent kinase (CDK) inhibitor roniciclib, the CDK4/6 inhibitor palbociclib, chemotherapy bendamustine, histone deacetylase (HDAC) inhibitors panobinostat and romidepsin, the immunomodulator (IMID) lenalidomide, the JAK1/2 inhibitor ruxolitinib, the MALT1 inhibitor MI2, the proteasome inhibitor bortezomib, and the PTEFb/CDK9 inhibitor atuveciclib. The anti-CD20 monoclonal antibody (moAb) rituximab was used only for B-cell lymphomas, whereas the ALK/MET inhibitor crizotinib and the anti-CD30 antibody drug conjugate brentuximab vedotin were only used in T-cell lymphomas.
Bortezomib, roniciclib, romidepsin, panobinostat, BAY 1238097, and atuveciclib were the most active in both B- and T-cell–derived cell lines, with median IC50 values lower than 1 μM (Table 2; supplemental Table 1). Other compounds with median IC50s in the nanomolar range were MI2 and ibrutinib in B-cell lymphomas (Table 2), and crizotinib in ALCL−ALK+ (median IC50, 57 nM; 95% CI, 32-100 nM). The remaining agents had median IC50s >1 μM (supplemental Table 1).
Approved antilymphoma agents and experimental inhibitors of key regulatory pathway with a median IC50 below 1 µM in B-cell and T-cell lymphoma cell lines
| Compound | IC50, nM | 95% CI, nM | Mechanism of action |
|---|---|---|---|
| B-cell lymphomas | |||
| Bortezomib | 5 | 5-7 | Proteasome inhibitor |
| Roniciclib | 23 | 18-29 | CDK inhibitor |
| Romidepsin | 34 | 2-94 | Class I HDAC inhibitor |
| Panobinostat | 161 | 11-1263 | Class I and II HDAC inhibitor |
| MI2 | 490 | 224-1000 | MALT1 inhibitor |
| BAY 1238097 | 684 | 463-1356 | BET inhibitor |
| Atuveciclib | 716 | 395-1020 | PTEFb/CDK9 inhibitor |
| Ibrutinib | 788 | 31-1000 | BTK inhibitor ibrutinib |
| T-cell lymphomas | |||
| Romidepsin | 2 | 2-5 | Class I HDAC inhibitor |
| Bortezomib | 3 | 2-6 | Proteasome inhibitor |
| Panobinostat | 10 | 5-14 | Class I and II HDAC inhibitor |
| Roniciclib | 21 | 16-42 | CDK inhibitor |
| BAY 1238097 | 410 | 282-690 | BET inhibitor |
| Atuveciclib | 567 | 324-1952 | PTEFb/CDK9 inhibitor |
| Compound | IC50, nM | 95% CI, nM | Mechanism of action |
|---|---|---|---|
| B-cell lymphomas | |||
| Bortezomib | 5 | 5-7 | Proteasome inhibitor |
| Roniciclib | 23 | 18-29 | CDK inhibitor |
| Romidepsin | 34 | 2-94 | Class I HDAC inhibitor |
| Panobinostat | 161 | 11-1263 | Class I and II HDAC inhibitor |
| MI2 | 490 | 224-1000 | MALT1 inhibitor |
| BAY 1238097 | 684 | 463-1356 | BET inhibitor |
| Atuveciclib | 716 | 395-1020 | PTEFb/CDK9 inhibitor |
| Ibrutinib | 788 | 31-1000 | BTK inhibitor ibrutinib |
| T-cell lymphomas | |||
| Romidepsin | 2 | 2-5 | Class I HDAC inhibitor |
| Bortezomib | 3 | 2-6 | Proteasome inhibitor |
| Panobinostat | 10 | 5-14 | Class I and II HDAC inhibitor |
| Roniciclib | 21 | 16-42 | CDK inhibitor |
| BAY 1238097 | 410 | 282-690 | BET inhibitor |
| Atuveciclib | 567 | 324-1952 | PTEFb/CDK9 inhibitor |
BTK, Bruton tyrosine kinase; CDK, cyclin-dependent kinase; HDAC, histone deacetylase; MALT1, mucosa-associated lymphoid tissue lymphoma translocation protein 1.
In B-cell lymphomas, various copanlisib-containing combinations showed synergistic inhibition of cell viability in vitro (Figure 1A). The most synergistic combination was copanlisib with venetoclax. We performed an in vivo experiment using the SSK41 MZL xenograft model to validate the beneficial effect of this combination and also of copanlisib plus lenalidomide, based on the IMID clinical activity as single agent or in combination in both MCL30 and MZL.31 Mice were treated with vehicle (IV), copanlisib (14 mg/kg IV, 2 days on/5 days off), venetoclax (200 mg/kg, postoperatively, once per day), lenalidomide (50 mg/kg, postoperatively, once per day), copanlisib plus venetoclax (same doses as the single agents), or copanlisib plus lenalidomide (same doses as the single agents). Both combination arms resulted in improved antitumor activity both vs the control and the individual arms (Figure 1C; supplemental Table 2). The copanlisib monotherapy was well tolerated with no body weight loss above 10% or loss of mice. The combination treatment showed substantial body weight losses in some animals, which was in the same range as for the monotherapies with venetoclax or lenalidomide (supplemental Figure 4).
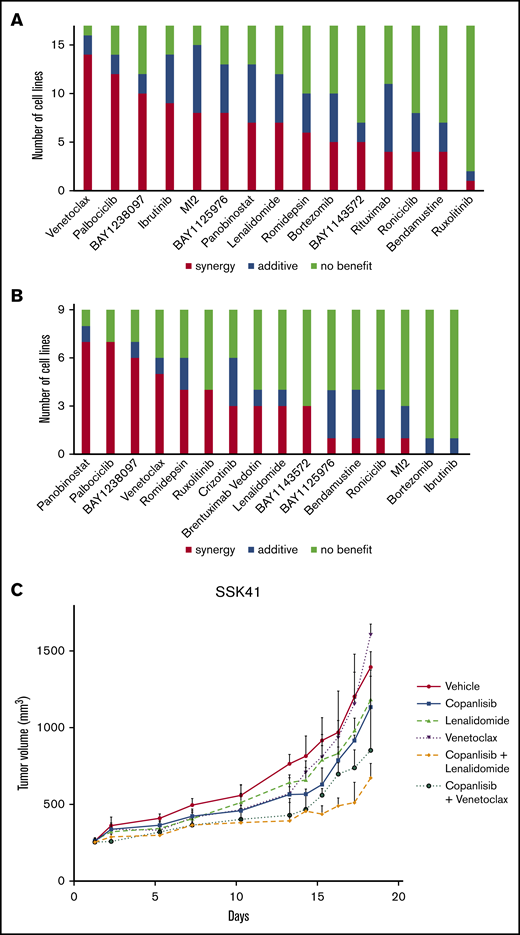
In vitro and in vivo antiproliferative effects of copanlisib-containing combinations in lymphoma models. (A) In vitro combination in B-cell lymphoma cell lines derived from MCL, MZL, and CLL. (B) In vitro combination in T-cell lymphomas cell lines derived from ALCL, PTCL, and CTCL. (C) Antitumor in vivo activity of copanlisib in combination with venetoclax or lenalidomide in the MZL SSK41 model. Mice were treated with vehicle (IV), copanlisib (14 mg kg IV, 2 days on/5 days off), venetoclax (200 mg/kg, postoperatively, once per day), lenalidomide (50 mg/kg, postoperatively, once per day), copanlisib plus venetoclax (same doses as the single agents), or copanlisib plus lenalidomide (same doses as the single agents). Lines show median values per time point with the corresponding upper interquartile range. The y-axis indicates the tumor volume in millimeters cubed; the x-axis, days of treatment (P values shown in supplemental Table 4).
In vitro and in vivo antiproliferative effects of copanlisib-containing combinations in lymphoma models. (A) In vitro combination in B-cell lymphoma cell lines derived from MCL, MZL, and CLL. (B) In vitro combination in T-cell lymphomas cell lines derived from ALCL, PTCL, and CTCL. (C) Antitumor in vivo activity of copanlisib in combination with venetoclax or lenalidomide in the MZL SSK41 model. Mice were treated with vehicle (IV), copanlisib (14 mg kg IV, 2 days on/5 days off), venetoclax (200 mg/kg, postoperatively, once per day), lenalidomide (50 mg/kg, postoperatively, once per day), copanlisib plus venetoclax (same doses as the single agents), or copanlisib plus lenalidomide (same doses as the single agents). Lines show median values per time point with the corresponding upper interquartile range. The y-axis indicates the tumor volume in millimeters cubed; the x-axis, days of treatment (P values shown in supplemental Table 4).
In T-cell lymphomas, the most beneficial combination was copanlisib with panobinostat, followed by copanlisib with palbociclib. Looking at specific T-cell subtypes, benefit was observed in all 4 ALCL−ALK+ with copanlisib/crizotinib (3 synergisms) (Figure 1B; supplemental Table 3). Conversely, all but 1 of the non–ALCL−ALK+ cell lines displayed synergy with copanlisib/ruxolitinib, which was very strong (combination index < 0.3) in the 2 SS-derived cell lines. Copanlisib/brentuximab vedotin was synergistic in the ALCL−ALK−, PTCL, and CTCL cell lines (supplemental Table 3).
The combination of copanlisib/venetoclax leads to apoptosis via downregulation of the antiapoptotic gene BCL-XL and MCL1
The combination of copanlisib with venetoclax was the most active in B-cell lymphomas and among the most synergistic combinations in T-cell lymphomas. The presence of the 2 drugs led to increased apoptosis as demonstrated by a dose-dependently increased PARP cleavage in 3 of 4 cell lines (2 MZL, 1 MZL) (Figure 2A), and by a reduction of living cells in primary cells derived from 3 MCL and 2 MZL patients (Figure 2B). The effect was maximal when the 2 compounds were concomitantly given (median combination index, 0.37; 95% CI, 0.23-0.69) and reduced when copanlisib preceded (median combination index, 0.58; 95% CI, 0.29-1.05) or followed venetoclax (median combination index, 0.87; 95% CI, 0.17-2.29), as shown by experiments performed in the MCL Jeko-1 cell line over a 48-hour period.
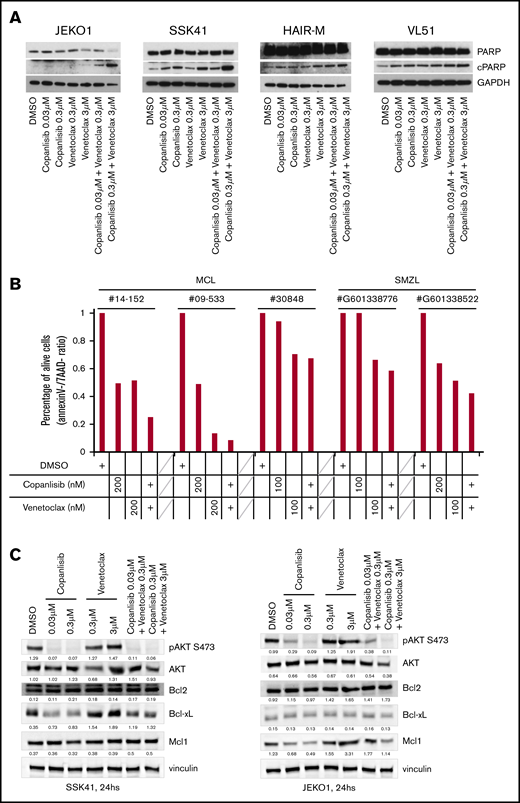
PI3K inhibitor copanlisib and the BCL2 inhibitor venetoclax are cytotoxic for MCL and MZL cells when combined and act via downregulating BCLXL or MCL1. (A) Four cell lines were exposed for 24 hours to copanlisib (30 nM, 300 nM), venetoclax (0.3 μM, 3 μM), or the combination of the 2 agents. (B) Three MCL and 2 MZL primary cells were exposed for 48 hours to copanlisib (100 or 200 nM), venetoclax (100 or 200 nM), or the combination of the 2 agents. The y-axis indicates the percentage of live cells, defined as Annexin V and 7AAD−, compared with dimethyl sulfoxide (DMSO)–treated cells. (C) Two cell lines exposed to copanlisib (30 nM, 300 nM), venetoclax (0.3 μM, 3 μM), or the combination of the 2 agents for 24 hours. cPARP, cleaved PARP; SMZL, splenic MZL.
PI3K inhibitor copanlisib and the BCL2 inhibitor venetoclax are cytotoxic for MCL and MZL cells when combined and act via downregulating BCLXL or MCL1. (A) Four cell lines were exposed for 24 hours to copanlisib (30 nM, 300 nM), venetoclax (0.3 μM, 3 μM), or the combination of the 2 agents. (B) Three MCL and 2 MZL primary cells were exposed for 48 hours to copanlisib (100 or 200 nM), venetoclax (100 or 200 nM), or the combination of the 2 agents. The y-axis indicates the percentage of live cells, defined as Annexin V and 7AAD−, compared with dimethyl sulfoxide (DMSO)–treated cells. (C) Two cell lines exposed to copanlisib (30 nM, 300 nM), venetoclax (0.3 μM, 3 μM), or the combination of the 2 agents for 24 hours. cPARP, cleaved PARP; SMZL, splenic MZL.
To understand the mechanisms underlying the synergism, we treated the MZL SSK41 and the MCL JEKO1 cell lines with copanlisib, venetoclax or a combination of the 2 drugs for 24 hours. Copanlisib treatment resulted in a dose-dependent downregulation of phosphorylated AKT in both cell lines and of antiapoptotic proteins (BCL-XL in MZL and MCL1 in MCL, respectively) (Figure 2C). These changes were maintained in cells exposed to the combination, but not seen after single BCL2 inhibitor treatment. No BCL2 protein expression changes were observed.
The combination of copanlisib/palbociclib leads to upregulation of PIK3IP1 and increased cell cycle arrest
The combination of copanlisib with palbociclib, was the second most active combination in both B-and T-cell lymphomas. CDK4/6 inhibitors are FDA approved for breast cancer.32,33 In lymphoma, promising preclinical and early clinical data, especially in MCL, have been reported.34-38 Therefore, this combination was explored in more detail. In contrast to the copanlisib/venetoclax combination, synergy was not due to apoptosis induction (Figure 3A) but to increased cell-cycle arrest in G0/G1 (Figure 3B). We observed an upregulation of PIK3IP1 in both transcriptome analysis of the copanlisib-treated HAIRM cells (logFC, 1.1; P = 1.86E-15; FDR = 6.57E-14) and, in real-time PCR analysis of 2 other different cell lines exposed to the combination of copanlisib and palbociclib (Figure 3C). The upregulation of PIK3IP1 has been shown to contribute to the synergism in MCL cell lines treated with another CDK4/6 inhibitor (PD0332991) and idelalisib.37
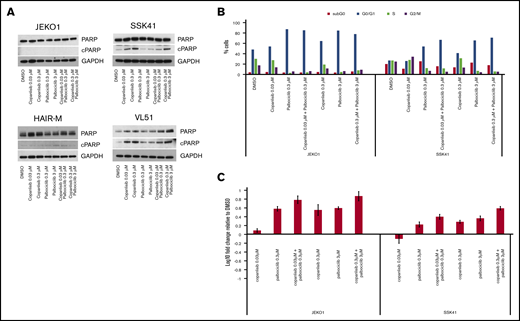
Combination of the PI3K inhibitor copanlisib with the CDK4/6 inhibitor palbociclib did not induce apoptosis in MCL and MZL cell lines, but did induce G0/G1 arrest with upregulation of PIK3IP1. (A) Four cell lines were exposed for 24 hours to copanlisib (30 nM, 300 nM), palbociclib (0.3 μM, 3 μM), or the combination of the 2 agents. (B) Cell-cycle analysis of 1 MCL (JEKO1) and 1 MZL (SSK41) cell line exposed for 24 hours to copanlisib (30 nM, 300 nM), palbociclib (0.3 μM, 3 μM), or the combination of the 2 agents. (C) RNA changes of PIK3IP1 in 1 MCL (JEKO1) and 1 MZL cell line exposed for 24 hours to copanlisib (30 nM, 300 nM), palbociclib (0.3 μM, 3 μM), or the combination of the 2 agents.
Combination of the PI3K inhibitor copanlisib with the CDK4/6 inhibitor palbociclib did not induce apoptosis in MCL and MZL cell lines, but did induce G0/G1 arrest with upregulation of PIK3IP1. (A) Four cell lines were exposed for 24 hours to copanlisib (30 nM, 300 nM), palbociclib (0.3 μM, 3 μM), or the combination of the 2 agents. (B) Cell-cycle analysis of 1 MCL (JEKO1) and 1 MZL (SSK41) cell line exposed for 24 hours to copanlisib (30 nM, 300 nM), palbociclib (0.3 μM, 3 μM), or the combination of the 2 agents. (C) RNA changes of PIK3IP1 in 1 MCL (JEKO1) and 1 MZL cell line exposed for 24 hours to copanlisib (30 nM, 300 nM), palbociclib (0.3 μM, 3 μM), or the combination of the 2 agents.
Transcriptome changes identify PIM and MCL1 inhibitors as additional partner for copanlisib
To assess the effects of copanlisib on the transcriptome, we exposed the MZL cell line HAIR-M to DMSO or copanlisib (5 nM, for 4, 8, and 12 hours). A total of 1924 and 2230 transcripts were downregulated and upregulated, respectively (supplemental Table 4A). The downregulated transcripts were enriched with genes involved in the MYC transcriptional program, mTOR signaling, glycolysis, response to unfolded protein, NF-κB signaling, and cancer cell essential genes (supplemental Figure 5; supplemental Table 4B-C). A significant enrichment for molecules involved in immune escape, including CD274, PDCD1, CSF1, and TIMP1, was observed. Upregulated transcripts were enriched with genes involved in IFNα signaling. Having previously reported gene expression signatures obtained with other signaling inhibitors in DLBCL cell lines,8 we compared these with the changes induced by copanlisib. An overlap of differentially expressed genes was observed with a PI3Kδ inhibitor, BTK inhibitor, AKT inhibitor, and dual inhibitors of PI3Kδ/PI3Kγ or mTOR/PI3K, with the exception that the HAIR-M did not show upregulation of genes involved in B-cell receptor signaling (supplemental Figure 6; supplemental Table 4B).
As the genes coding for the PIM kinases (PIM2, log2FC = 1.3, adjusted P = 1.25E-22; PIM1, log2FC = 0.6, adjusted P = 3.78E-14) and the antiapoptotic gene MCL1 (log2FC = 0.5; adjusted P = 7.69E-18) were upregulated (supplemental Table 2A), we combined copanlisib with the PIM inhibitor AZD1208 and the MCL1 inhibitor MIK665 (S63845) in 3 MCL and 3 MZL cell lines. Both combinations were synergistic in HAIR-M and beneficial in additional 5 of 5 and 3 of 5 cell lines, respectively (Table 3).
In vitro assessment of copanlisib combination with PIM or MCL1 inhibitors
| Target | Combination partner | Histology | Cell line | Median combination index | 95% CI |
|---|---|---|---|---|---|
| PIM1/2 | AZD1208 | MZL | ESKOL | 0.79 | 0.62-1.10 |
| PIM1/2 | AZD1208 | MZL | HAIR-M | 0.62 | 0.35-1.11 |
| PIM1/2 | AZD1208 | MZL | HC1 | 0.23 | 0.14-0.51 |
| PIM1/2 | AZD1208 | MCL | MAVER1 | 0.99 | 0.59-1.88 |
| PIM1/2 | AZD1208 | MCL | MINO | 0.60 | 0.46-0.85 |
| PIM1/2 | AZD1208 | MCL | Z138 | 0.84 | 0.56-1.23 |
| MCL1 | MIK665 | MZL | ESKOL | 1.25 | 1.13-1.58 |
| MCL1 | MIK665 | MZL | HAIR-M | 0.65 | 0.29-1.36 |
| MCL1 | MIK665 | MZL | HC1 | 0.24 | 0.18-.67 |
| MCL1 | MIK665 | MCL | MAVER1 | >3 | n.d. |
| MCL1 | MIK665 | MCL | MINO | 0.67 | 0.41-1.27 |
| MCL1 | MIK665 | MCL | Z138 | 0.9 | 0.59-1.17 |
| Target | Combination partner | Histology | Cell line | Median combination index | 95% CI |
|---|---|---|---|---|---|
| PIM1/2 | AZD1208 | MZL | ESKOL | 0.79 | 0.62-1.10 |
| PIM1/2 | AZD1208 | MZL | HAIR-M | 0.62 | 0.35-1.11 |
| PIM1/2 | AZD1208 | MZL | HC1 | 0.23 | 0.14-0.51 |
| PIM1/2 | AZD1208 | MCL | MAVER1 | 0.99 | 0.59-1.88 |
| PIM1/2 | AZD1208 | MCL | MINO | 0.60 | 0.46-0.85 |
| PIM1/2 | AZD1208 | MCL | Z138 | 0.84 | 0.56-1.23 |
| MCL1 | MIK665 | MZL | ESKOL | 1.25 | 1.13-1.58 |
| MCL1 | MIK665 | MZL | HAIR-M | 0.65 | 0.29-1.36 |
| MCL1 | MIK665 | MZL | HC1 | 0.24 | 0.18-.67 |
| MCL1 | MIK665 | MCL | MAVER1 | >3 | n.d. |
| MCL1 | MIK665 | MCL | MINO | 0.67 | 0.41-1.27 |
| MCL1 | MIK665 | MCL | Z138 | 0.9 | 0.59-1.17 |
Cell lines were exposed to increasing concentrations of copanlisib and/or of the other compound for 72 hours.
Discussion
After the first demonstration of clinical activity of copanlisib in patients with lymphomas,16 we started a project to identify novel active combinations especially for non DLBCL patients. With this aim, we studied copanlisib as a single agent and in combination with 17 additional drugs in a panel of 26 B-cell or T-cell lymphomas. Copanlisib as a single agent showed an in vitro dose-dependent antiproliferative activity in a variety of models derived from MCL, MZL, and T-cell lymphomas. These data were confirmed in vivo in MZL and MCL xenografts, extend the data reported in DLBCL,7,10 and provide a strong biological basis for the results obtained in the clinical setting.17,18
The IC50 values for copanlisib were well within the concentrations that are clinically achievable (∼180-190 nM)16 for the vast majority of cell lines and in particular for all the B-cell lymphoma models. The lower activity observed in the cell lines derived from T-cell lymphomas is in line with previous results using other PI3K or dual PI3K/mTOR inhibitors.8,39-41 When considering B- and T-cell lymphoma separately, no correlations were observed between sensitivity to copanlisib and PI3K isoforms. Across all cell lines, copanlisib presented a pattern of activity that was similar to what we have previously observed with the PI3Kδ inhibitor idelalisib and the dual PI3K/mTOR inhibitors bimiralisib and apitolisib,8 but copanlisib was significantly more potent than the other compounds.
The combination screen identified several compounds that synergized with copanlisib in vitro. The strongest combination was with BCL2 inhibitor venetoclax. The benefit of the combination over single agent was also validated in an MZL xenograft model and in MCL and MZL primary cells, and was due to increased induction of apoptosis, likely sustained by the reduction of the antiapoptotic proteins MCL1 and BCL-XL, observed in MCL and MZL cell lines, respectively. These data provided the rational for the design of the SAKK 66/18 phase 1 study currently exploring the combination of copanlisib and venetoclax in relapsed/refractory lymphomas. Based on the toxicity profiles of the single agents,16-22,42,43 there are potential overlapping side effects, such as neutropenia, infections, diarrhea, nausea, vomiting, and fatigue, which will be assessed in the clinical study. Our data are in agreement with what reported in DLBCL preclinical models,10 and with venetoclax combined with other PI3K inhibitors.8,10,44,45
The observed benefit of the combination with the BTK-inhibitor ibrutinib was in agreement with what was previously reported in the DLBCL context7 and also with data obtained with BTK inhibitors plus other PI3K inhibitors.8,44,46-49 We did not further studied this combination here, but there are 2 ongoing phase 1/2 studies looking at the combination in refractory/recurrent MCL (NCT03877055) and in refractory/recurrent primary central nervous system lymphoma (NCT03581942).
The combination with the IMID lenalidomide was validated in vivo in an MZL model. Considering the clinical activity of both agents in the MZL and MCL context,30,31 the combination looks promising for further clinical investigations, although its potential toxicity must be carefully considered due to lethal events observed in combinations of lenalidomide with the PI3Kδ inhibitor idelalisib plus the anti-CD20 rituximab.50
The CDK4/6 inhibitors ribociclib and palbociclib are FDA approved for breast cancer.32,33 In our study the addition of palbociclib to copanlisib represented to the second-best combination both in B- and T-cell lymphoma cell lines. In contrast to copanlisib plus venetoclax, this combination was associated with a stronger cell-cycle arrest but no increased cytotoxicity compared with the drugs as single agents. Indeed, we observed higher expression levels of PIK3IP1, similarly to the effect of adding the CDK4 inhibitor to idelalisib or ibrutinib in MCL cells.36,37
Among T-cell lymphomas, copanlisib plus the class I and IIa/b HDAC inhibitor panobinostat was the most active combination. Albeit at lesser extent, the class I HDAC inhibitor romidepsin was beneficial as well when added to copanlisib, and this combination is the subject of a very recently designed phase 1 study for refractory/recurrent mature T-cell lymphomas (NCT04233697, not yet recruiting on 30 January 2020). Importantly, HDAC inhibitors have recognized antitumor activity in PTCL and/or CTCL (romidepsin, belinostat, and vorinostat are FDA approved),51 and previous preclinical and early clinical data confirm the benefit of inhibiting both HDAC and PI3K pathway.52
Exposure to copanlisib in an MZL cell line heavily affected the lymphoma cell transcriptome, downregulating the MYC transcriptional program, alongside NF-κB signaling, mTOR signaling, glycolysis, genes that are essential for cancer cells and also molecules important for immune escape (CD274, PDCD1, CSF1, TIMP1), suggesting the exploration of combinations including copanlisib and immune-checkpoint modulators. Since we observed an upregulation of PIM1 and PIM2 kinases, similarly to what we observed after dual PI3K/mTOR pharmacological inhibition,8 the addition of a PIM inhibitor to copanlisib was beneficial in all the 6 MCL and MZL cell lines tested. Adding an MCL1 inhibitor to copanlisib appeared beneficial in 4 of 6 cell lines. Indeed, MCL1 differs from PIM1 and PIM2 and is not always upregulated by copanlisib, but as seen in the JEKO1 MCL cell line or in DLBCL cell lines,10 it can be directly inactivated by the PI3K inhibitor, suggesting this combination is not required.
Our study describes the antitumor activity as single agents of different drugs in MZL, MCL and T-cell lymphoma models. While the strong antiproliferative activity of the proteasome inhibitor bortezomib, the HDAC and BET inhibitors, the MALT1 inhibitor MI2 were expected, our data represent novel findings for other compounds. The CDK inhibitor roniciclib appeared equally active in lymphoma cell lines compared with solid tumor models.53 So far, only few clinical data are available for lymphomas, with 3 stable diseases among 7 patients with unspecified lymphoid tumors in a phase 1 study.54 Another compound with single agent activity was the PTEFb/CDK9 inhibitor atuveciclib, which has already shown preclinical antitumor activity in adult T-cell leukemia/lymphoma55 and acute myeloid leukemia56 and has entered the phase 1 study for patients with acute myeloid leukemia (NCT02345382).
In conclusion, we identified novel copanlisib combination partners with increased antineoplastic activity in different lymphoma models. In particular, data presented here provided the rationale for a phase 1 study exploring the combination of copanlisib with venetoclax (SAKK 66/18, NCT03886649).
Profiling data are available at the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) database under accession number GSE137325.
Acknowledgments
This work was supported by institutional research funds from Bayer AG (Berlin, Germany), research grants of the Barletta and Gelu Foundations (F.B.), and Swiss Cancer Research KLS-3636-02-2015 (A.R.).
Authorship
Contribution: C.T., M.L., and E.G. designed and performed experiments, interpreted data, and wrote the manuscript; I.K., L.C., and F.B. performed data mining; F.S., T.J., M.B., A. Sturz, A.J.A., C.S., F.M., and L.S. performed experiments; A.R. performed transcriptome profiling; G.G. provided clinical specimens; A. Stathis, D.R., N.L., and E.Z. provided advice; O.P. designed the study, interpreted data, and wrote the manuscript; F.B. designed the study, performed data mining, interpreted data, supervised the study, and wrote the manuscript; and all authors approved the final manuscript.
Conflict-of-interest disclosure: M.L., T.J., M.B., A. Sturz, C.S., N.L., and O.P. are employees of Bayer AG. L.C. received a travel grant from HTG. D.R. received grant support from Gilead, AbbVie, and Janssen; honoraria from Gilead, AbbVie, Janssen, and Roche; and scientific advisory board fees from Gilead, AbbVie, Janssen, AstraZeneca, and MSD. A. Stathis received institutional research funds from Bayer, ImmunoGen, Merck, Pfizer, Novartis, and Roche; and a travel grant from AbbVie. E.Z. received institutional research funds from Celgene, Roche, and Janssen; received advisory board fees from Celgene, Roche, Mei Pharma, AstraZeneca, and Celltrion Healthcare; received travel grants from AbbVie and Gilead; and provided expert statements to Gilead, Bristol-Myers Squibb, and MSD. F.B. received institutional research funds from Acerta, ADC Therapeutics, Bayer AG, Cellestia, CTI Life Sciences, EMD Serono, Helsinn, ImmunoGen, Menarini Ricerche, NEOMED Therapeutics 1, Nordic Nanovector ASA, Oncology Therapeutic Development, and PIQUR Therapeutics AG; received consultancy fees from Helsinn and Menarini; provided expert statements to HTG; and received travel grants from Amgen, AstraZeneca, Jazz Pharmaceuticals, and PIQUR Therapeutics AG. The remaining authors declare no competing financial interests.
Correspondence: Francesco Bertoni, Institute of Oncology Research, Università della Svizzera Italiana, via Vincenzo Vela 6, 6500 Bellinzona, Switzerland; e-mail: francesco.bertoni@ior.usi.ch.

