

Key Points
Circulating CLL fractions share a dominant intraclonal DNA methylation hallmark that reflects cell-of-origin and IGHV mutation of leukemia.
Intraclonal epigenetic and transcriptional changes support a progressive life cycle of CLL that is potentially connected to clinical outcome.

Abstract
Intraclonal subpopulations of circulating chronic lymphocytic leukemia (CLL) cells with different proliferative histories and reciprocal surface expression of CXCR4 and CD5 have been observed in the peripheral blood of CLL patients and named proliferative (PF), intermediate (IF), and resting (RF) cellular fractions. Here, we found that these intraclonal circulating fractions share persistent DNA methylation signatures largely associated with the mutation status of the immunoglobulin heavy chain locus (IGHV) and their origins from distinct stages of differentiation of antigen-experienced B cells. Increased leukemic birth rate, however, showed a very limited impact on DNA methylation of circulating CLL fractions independent of IGHV mutation status. Additionally, DNA methylation heterogeneity increased as leukemic cells advanced from PF to RF in the peripheral blood. This frequently co-occurred with heterochromatin hypomethylation and hypermethylation of Polycomb-repressed regions in the PF, suggesting accumulation of longevity-associated epigenetic features in recently born cells. On the other hand, transcriptional differences between paired intraclonal fractions confirmed their proliferative experience and further supported a linear advancement from PF to RF in the peripheral blood. Several of these differentially expressed genes showed unique associations with clinical outcome not evident in the bulk clone, supporting the pathological and therapeutic relevance of studying intraclonal CLL fractions. We conclude that independent methylation and transcriptional landscapes reflect both preexisting cell-of-origin fingerprints and more recently acquired hallmarks associated with the life cycle of circulating CLL cells.
Introduction
Chronic lymphocytic leukemia (CLL), the most common adult leukemia in the western world, is characterized by progressive accumulation of immunophenotypically distinct CD5+ lymphocytes.1,2 Clinical staging systems by Rai3 and Binet4 are partially based on the accumulation of CLL cells in lymphoid tissues, highlighting the relevance of leukemic cell homing mechanisms. Indeed, the study of surface membrane molecules, including chemokine receptor 4 (CXCR4), and in vivo measurements of proliferation, based on deuterium (2H)-labeling of dividing CLL cells, showed that the peripheral blood contains intraclonal cellular fractions with different trafficking potentials and proliferative histories.5,6 Specifically, differential surface densities of CXCR4 and CD5 suggested a heterogeneous continuum of CLL cells from those that had recently divided and migrated out the lymphoid tissues into the circulation (CXCR4DimCD5Bright; proliferative fraction [PF]) to older cells with increased expression of CXCR4 (CXCR4BrightCD5Dim; resting fraction [RF]) that may be attempting to home back to solid tissues. This is consistent with the finding that the impairment of signaling and expression of CXCR4 by the Bruton tyrosine kinase inhibitor ibrutinib7,8 promotes the mobilization of CLL cells into the peripheral circulation and blocks their homing to solid tissues.9,10
Molecularly, CLL patients can be subdivided into 2 subsets with distinct clinical and biological characteristics based on the presence or absence of somatic mutations in the variable region of the immunoglobulin heavy chain (IGHV).1,11 Compared with patients with IGHV mutations (M-CLL), IGHV-unmutated patients (U-CLL) have a more aggressive disease course that is more rapidly fatal without treatment.12,13 Additional molecular profiling of CLL has revealed not only complex genetic interpatient differences14-17 but also epigenetic differences in chromatin landscape18,19 and DNA methylation,20 reflecting complex molecular and microenvironmental interactions during the development and evolution of CLL.2,21-23 Specifically, global DNA methylation patterns can predict distinct CLL epigenetic subtypes with prognostic implications, possibly reflecting their cellular origin, IGHV mutational experience, and their relationship to later stages of normal B-cell development.20,24-26 In addition, more aggressive disease progression is often associated with high leukemic birth rates (BRs)27,28 and intraclonal genetic heterogeneity,29,30 indicating that clonal evolution is a key factor in the disease. Likewise, further evolution of DNA methylation may occur in high-risk clinically progressive cases, coevolving with genetic aberrations.31 Such somatic epigenetic heterogeneity in CLL has been shown to accumulate intraclonally, potentially facilitating the accrual of additional subclonal mutations and promoting shorter remission times after treatment.32 It remains unknown whether molecular heterogeneity might also correlate with BR of CLL cells in vivo or contribute to the appearance of the observed CXCR4/CD5 intraclonal subpopulations.
To address these questions, we determined DNA methylation and gene expression changes occurring within each of the more homogeneous intraclonal CLL fractions, which were highly enriched in recently born or in older quiescent leukemic cells. Patient samples were obtained independently from a subgroup of patients who participated in the CRC011 Heavy Water CLL Research Consortium trial,28 which examined the usefulness of leukemic cell BR in the prognosis of CLL. This allowed us to associate the epigenetic profiling of CLL fractions with patient BR, clinical outcome, and typical biomarkers of prognosis. Studying sorted intraclonal subpopulations provided us the opportunity to examine the extent to which the fractions reflect cyclic events occurring in circulating leukemic cells that could reveal differences possibly obscured by the heterogeneity of the bulk CLL clone.
Methods
Patients
Twenty-one previously untreated early-stage (Rai stage 0, 1, or 2) patients with CLL followed at the Northwell Health Cancer Institute and The James Cancer Center, Ohio State University who participated in the CRC011 Heavy Water trial (NCT00481858)28 were studied (Table 1). This cohort of 8 U-CLL and 13 M-CLL cases was selected based on additional sample material availability and in general was a good representation of the overall trial cohort (Figure 1). Expression of ZAP70 and CD38, IGHV mutational status, and leukemic BRs of florescence-activated cell sorter (FACS)-sorted CD19+CD5+ peripheral cells were extracted from the published findings.28
Clinical and molecular characteristics of CLL patients in this study
| Patient | Age, y | Sex | Rai stage | IGHV mutation | Methylation | Immunophenotype | 2H2O | Cytogenetic abnormalities, % cells | Outcome | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Diff | Status | 5 CpGs | ZAP70, % cells | CD38, % cells | BR, %/d | Δ17p23 | Δ13q14 | Tri12 | Δ11q22 | Trt | TTFT, mo | ||||
| P40489 | 51 | M | 0 | 7.0 | M-CLL | m-CLL | 3.92 | 1.1 | 0.14 | 0 | 0 | 0 | 0 | — | 22 |
| CLL606 | 49 | M | 0 | 6.2 | M-CLL | m-CLL | 1.7 | 0.4 | 0.15 | 0 | 96 | 0 | 0 | — | 76 |
| CLL1026 | 44 | F | 0 | 10.3 | M-CLL | m-CLL | 1.4 | 2.1 | 0.16 | 0 | 0 | 0 | 0 | — | 45 |
| CLL276 | 68 | M | 1 | 5.3 | M-CLL | m-CLL | 4.3 | 6.6 | 0.19 | ND | ND | ND | ND | BR | 23 |
| CLL1127 | 60 | M | 0 | 7.6 | M-CLL | m-CLL | 0.42 | 3.4 | 0.21 | 0 | 84 | 0 | 0 | — | 60 |
| CLL373 | 52 | F | 1 | 12.4 | M-CLL | m-CLL | 16.8 | 1.2 | 0.28 | ND | ND | ND | ND | — | 58 |
| CLL584 | 53 | M | 0 | 6.6 | M-CLL | m-CLL | 1 | 1.2 | 0.28 | 0 | 0 | 0 | 0 | — | 75 |
| P40323 | 63 | F | 0 | 9.8 | M-CLL | m-CLL | 7.34 | 1.7 | 0.29 | 2 | 71.5 | 0 | 0 | — | 39 |
| CLL638 | 53 | M | 0 | 3.2 | M-CLL | n-CLL | 63.7 | 0.8 | 0.31 | 0 | 0 | 0 | 0 | — | 58 |
| P40344 | 55 | M | 1 | 6.6 | M-CLL | m-CLL | 19 | 9.1 | 0.35 | 0 | 0 | 0 | 0 | O | 31 |
| CLL331 | 77 | M | 1 | 3.9 | M-CLL | m-CLL | 73.6 | 11 | 0.36 | ND | ND | ND | ND | — | 69 |
| CLL1039 | 51 | F | 0 | 3.9 | M-CLL | m-CLL | 1.7 | 0.9 | 0.37 | 0 | 88 | 0 | 0 | FCR ×4 | 9 |
| CLL499 | 65 | M | 2 | 6.0 | M-CLL | m-CLL | 0.9 | 3.3 | 0.58 | ND | ND | ND | ND | FR ×4 | 9 |
| CLL650 | 53 | M | 1 | 0.0 | U-CLL | i-CLL | 40.2 | 94.9 | 0.20 | 0 | 0 | 0 | 69.5 | FR ×2 | 10 |
| CLL609 | 61 | M | 1 | 0.3 | U-CLL | n-CLL | 51.3 | 16.8 | 0.33 | 0 | 11.5 | 0 | 0 | FCR ×6 | 28 |
| CLL625 | 53 | M | 2 | 0.0 | U-CLL | n-CLL | 32.3 | 41.7 | 0.39 | 0 | 0 | 0 | 100 | FR | 6 |
| CLL452 | 51 | M | 1 | 0.0 | U-CLL | n-CLL | 43.8 | 20.5 | 0.48 | ND | ND | ND | ND | FR | 16 |
| CLL934 | 57 | F | 1 | 0.0 | U-CLL | n-CLL | 61.1 | 4.5 | 0.56 | 0 | 0 | 53.5 | 0 | FR ×5 | 7 |
| CLL569 | 55 | M | 2 | 0.7 | U-CLL | n-CLL | 45.8 | 86.5 | 0.81 | 0 | 0 | 0 | 0 | FCR | 16 |
| CLL493 | 56 | M | 2 | 0.0 | U-CLL | n-CLL | 93.8 | 98 | 0.96 | ND | ND | ND | ND | RCD ×3 | 19 |
| CLL753 | 69 | M | 1 | 0.0 | U-CLL | n-CLL | 73.5 | 76.3 | 1.03 | ND | ND | ND | ND | FCR ×4 | 2 |
| Patient | Age, y | Sex | Rai stage | IGHV mutation | Methylation | Immunophenotype | 2H2O | Cytogenetic abnormalities, % cells | Outcome | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Diff | Status | 5 CpGs | ZAP70, % cells | CD38, % cells | BR, %/d | Δ17p23 | Δ13q14 | Tri12 | Δ11q22 | Trt | TTFT, mo | ||||
| P40489 | 51 | M | 0 | 7.0 | M-CLL | m-CLL | 3.92 | 1.1 | 0.14 | 0 | 0 | 0 | 0 | — | 22 |
| CLL606 | 49 | M | 0 | 6.2 | M-CLL | m-CLL | 1.7 | 0.4 | 0.15 | 0 | 96 | 0 | 0 | — | 76 |
| CLL1026 | 44 | F | 0 | 10.3 | M-CLL | m-CLL | 1.4 | 2.1 | 0.16 | 0 | 0 | 0 | 0 | — | 45 |
| CLL276 | 68 | M | 1 | 5.3 | M-CLL | m-CLL | 4.3 | 6.6 | 0.19 | ND | ND | ND | ND | BR | 23 |
| CLL1127 | 60 | M | 0 | 7.6 | M-CLL | m-CLL | 0.42 | 3.4 | 0.21 | 0 | 84 | 0 | 0 | — | 60 |
| CLL373 | 52 | F | 1 | 12.4 | M-CLL | m-CLL | 16.8 | 1.2 | 0.28 | ND | ND | ND | ND | — | 58 |
| CLL584 | 53 | M | 0 | 6.6 | M-CLL | m-CLL | 1 | 1.2 | 0.28 | 0 | 0 | 0 | 0 | — | 75 |
| P40323 | 63 | F | 0 | 9.8 | M-CLL | m-CLL | 7.34 | 1.7 | 0.29 | 2 | 71.5 | 0 | 0 | — | 39 |
| CLL638 | 53 | M | 0 | 3.2 | M-CLL | n-CLL | 63.7 | 0.8 | 0.31 | 0 | 0 | 0 | 0 | — | 58 |
| P40344 | 55 | M | 1 | 6.6 | M-CLL | m-CLL | 19 | 9.1 | 0.35 | 0 | 0 | 0 | 0 | O | 31 |
| CLL331 | 77 | M | 1 | 3.9 | M-CLL | m-CLL | 73.6 | 11 | 0.36 | ND | ND | ND | ND | — | 69 |
| CLL1039 | 51 | F | 0 | 3.9 | M-CLL | m-CLL | 1.7 | 0.9 | 0.37 | 0 | 88 | 0 | 0 | FCR ×4 | 9 |
| CLL499 | 65 | M | 2 | 6.0 | M-CLL | m-CLL | 0.9 | 3.3 | 0.58 | ND | ND | ND | ND | FR ×4 | 9 |
| CLL650 | 53 | M | 1 | 0.0 | U-CLL | i-CLL | 40.2 | 94.9 | 0.20 | 0 | 0 | 0 | 69.5 | FR ×2 | 10 |
| CLL609 | 61 | M | 1 | 0.3 | U-CLL | n-CLL | 51.3 | 16.8 | 0.33 | 0 | 11.5 | 0 | 0 | FCR ×6 | 28 |
| CLL625 | 53 | M | 2 | 0.0 | U-CLL | n-CLL | 32.3 | 41.7 | 0.39 | 0 | 0 | 0 | 100 | FR | 6 |
| CLL452 | 51 | M | 1 | 0.0 | U-CLL | n-CLL | 43.8 | 20.5 | 0.48 | ND | ND | ND | ND | FR | 16 |
| CLL934 | 57 | F | 1 | 0.0 | U-CLL | n-CLL | 61.1 | 4.5 | 0.56 | 0 | 0 | 53.5 | 0 | FR ×5 | 7 |
| CLL569 | 55 | M | 2 | 0.7 | U-CLL | n-CLL | 45.8 | 86.5 | 0.81 | 0 | 0 | 0 | 0 | FCR | 16 |
| CLL493 | 56 | M | 2 | 0.0 | U-CLL | n-CLL | 93.8 | 98 | 0.96 | ND | ND | ND | ND | RCD ×3 | 19 |
| CLL753 | 69 | M | 1 | 0.0 | U-CLL | n-CLL | 73.5 | 76.3 | 1.03 | ND | ND | ND | ND | FCR ×4 | 2 |
BR, bendamustine, rituximab; F, female; FCR, fludarabine, cyclophosphamide, rituximab; FR, fludarabine, rituximab; i-CLL, intermediate CLL; M, male; m-CLL, memory B-cell–like CLL; M-CLL, IGHV-mutated CLL; n-CLL, naive-cell–like CLL; ND, not determined; O, ofatumumab; RCD, rituximab, cyclophosphamide, dexamethasone; Trt, treatment required within a 7-year follow-up period (numbers indicate cycles of treatment, if received); TTFT, time to first treatment (or last follow-up if untreated) from the start time of the heavy water protocol; U-CLL, IGHV-unmutated CLL; % Diff, percent deviation to germline in IGHV sequence (<2% is considered unmutated).
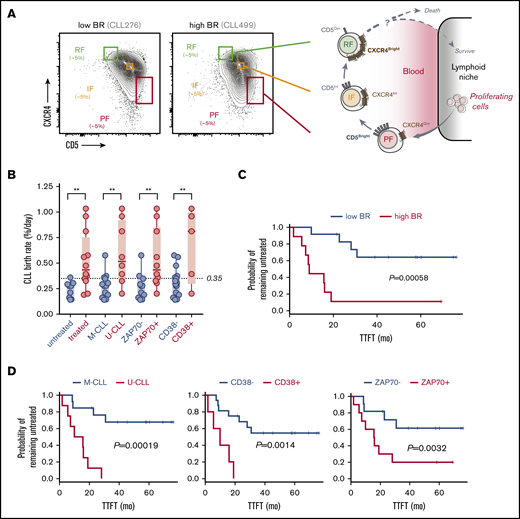
Leukemic-cell birth associates with other poor prognostic factors in CLL patients who carry circulating CXCR4/CD5 leukemic fractions. (A) Cytometry profiles of 2 representative CLL patients with low and high BR illustrating the inverse surface expression of CXCR4 and CD5 in the gating strategy that was used for all of the patients (left). Proposed model of the CLL life cycle, where differential surface densities of CXCR4 and CD5 identify 3 intraclonal fractions from cells that have recently divided and egressed from the lymphoid niche (CXCR4DimCD5Bright; PF) to an IF more representative of the bulk CLL clone to older quiescent cells that may be attempting to home back and escape cell death (CXCR4BrightCD5Dim; RF) (right). (B) Association of cellular BRs with different CLL prognostic factors, including the requirement of treatment during the study period (0-7 years), IGHV mutational status, and levels of surface ZAP70 or CD38 in the 21 CLL patients studied here. Unpaired nonparametric Mann-Whitney U tests were used to explore differences. Circles represent CLL patients in each clinical subcategory. **P < .01. (C) Kaplan-Meier curves of TTFT for CLL patients stratified according to low or high BR (dichotomized at 0.35% per day). (D) Kaplan-Meier curves of TTFT for CLL patients stratified according to different prognostic factors. P values in panels C and D were determined using the log-rank test.
Leukemic-cell birth associates with other poor prognostic factors in CLL patients who carry circulating CXCR4/CD5 leukemic fractions. (A) Cytometry profiles of 2 representative CLL patients with low and high BR illustrating the inverse surface expression of CXCR4 and CD5 in the gating strategy that was used for all of the patients (left). Proposed model of the CLL life cycle, where differential surface densities of CXCR4 and CD5 identify 3 intraclonal fractions from cells that have recently divided and egressed from the lymphoid niche (CXCR4DimCD5Bright; PF) to an IF more representative of the bulk CLL clone to older quiescent cells that may be attempting to home back and escape cell death (CXCR4BrightCD5Dim; RF) (right). (B) Association of cellular BRs with different CLL prognostic factors, including the requirement of treatment during the study period (0-7 years), IGHV mutational status, and levels of surface ZAP70 or CD38 in the 21 CLL patients studied here. Unpaired nonparametric Mann-Whitney U tests were used to explore differences. Circles represent CLL patients in each clinical subcategory. **P < .01. (C) Kaplan-Meier curves of TTFT for CLL patients stratified according to low or high BR (dichotomized at 0.35% per day). (D) Kaplan-Meier curves of TTFT for CLL patients stratified according to different prognostic factors. P values in panels C and D were determined using the log-rank test.
Isolation of cell fractions
Peripheral blood mononuclear cells were separated by Ficoll-Paque density gradient centrifugation from heparinized venous blood obtained 3 months before, during, or 3 months after the 6-week 2H2O labeling period and cryopreserved. Before sorting, CLL peripheral blood mononuclear cells were thawed and incubated with the murine anti-human monoclonal antibodies CD5-PE-Cy7, CXCR4-PE, and CD19-Pacific Blue (BD Biosciences). After gating on CD19+CD5+ cells, equal numbers of viable cells falling into the PF, intermediate (CXCR4IntCD5Int; intermediate fraction [IF]), and RF based on reciprocal densities of CXCR4 and CD56 (∼5% of the clone for each fraction) were sorted with a BD FACSAria (Figure 1A). After sorting, cells were washed, pelleted, and stored at −80° until DNA or RNA extraction was performed.
DNA methylation profiling
We used the EZ DNA Methylation kit (Zymo Research) for bisulfite conversion of 500 ng of genomic DNA obtained from at least 0.5 × 106 cells of each fractionated CLL sample. Subsequently, methylation high-density profiling was carried out by using the Illumina Infinium HD Human Methylation 450K BeadChip according to manufacturer’s instructions. See supplemental Methods for additional information regarding DNA methylation analysis and annotation.
Gene expression profiling
We extracted total RNA from sorted cells using the RNeasy mini kit (Qiagen) following manufacturer’s recommendations and assessed RNA quality with a Bioanalyzer (Agilent) using Agilent RNA 6000 Kit. We hybridized RNA samples to the Illumina HumanHT-12 v4 Expression BeadChip following Illumina standard protocols. See supplemental Methods for additional information regarding gene expression analysis and annotation.
Time to first treatment (TTFT) analysis
TTFT was calculated according to CRC011 trial28 from the date of the beginning of heavy water treatment to clinical progression, censored at the end of the follow-up period of 7 years or at the time of exclusion of patients from the study for non–treatment-related reasons. See supplemental Methods for additional information regarding Cox proportional hazard and log-rank tests evaluated with TTFT.
Results
High leukemic BRs associate with poor outcome predictors in patients with CXCR4/CD5 intraclonal CLL fractions
We analyzed cryopreserved cells from 21 of 97 recently diagnosed, untreated CLL patients (Table 1) who participated in the CRC011 Heavy Water clinical trial.28 To gain further insight into the origin, progression, and life cycle of CLL, we further subdivided the leukemic CD19+CD5+ population (total fraction) into CXCR4DimCD5Bright (PF), CXCR4IntCD5Int (IF), and CXCR4BrightCD5Dim (RF) fractions. The relationship of these fractions to the previously proposed life cycle of CLL cells5,6 is shown in Figure 1A. We dichotomized these patients (8 U-CLL and 13 M-CLL) using 0.35% cells/day (Figure 1B) as the established threshold between fast and slow BRs.27,28 This patient subset had clinical and genetic characteristics that were representative of this trial. Specifically, these patients showed significant association of high BR with several previously established predictors of disease progression requiring early treatment (increased numbers of ZAP70+ and CD38+ cells and unmutated IGHV status) (Figure 1B; Table 1). Furthermore, log-rank tests showed that BR (Figure 1C), IGHV mutation status, and percentage of CD38+ or ZAP70+ B cells (Figure 1D) were significantly associated with outcome as expected, supporting that our patient cohort generally extrapolates to the larger trial.
A dominant IGHV mutation–associated DNA methylation signature is stable in all intraclonal CLL fractions
To explore DNA methylation dynamics throughout the intraclonal life cycle of circulating CLL cells, we compared the 3 fractions to unfractionated (total/bulk) CLL cells (analyzed in most studies) using high-density DNA methylation microarrays.33 Unsupervised hierarchical clustering (HC) of the methylomes revealed that fractions uniformly clustered by patient (Figure 2A), suggesting strong intraclonal epigenetic similarities. In addition, both HC and principal component analysis (PCA) (Figure 2B) revealed that the clearest differences existed between M-CLL and U-CLL patients. Such patient-specific clustering persisted even after focusing on the top (∼5%) most variable cytosine guanine dinucleotides (CpGs) (supplemental Figure 1A).
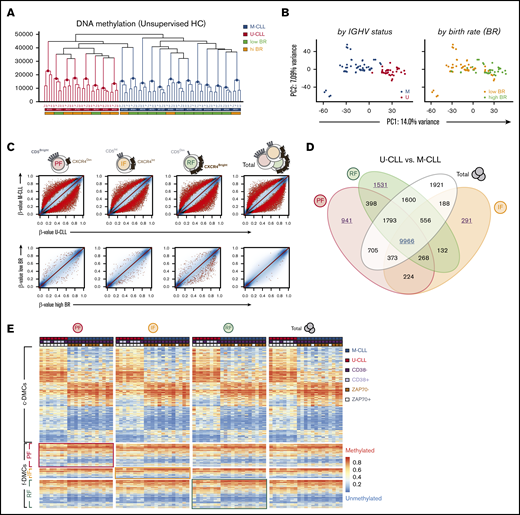
Intraclonally stable methylation patterns in CLL reflect signatures of the potential normal B-cell of origin. (A) Unsupervised HC of the fractions by Manhattan distance measurement of DNA methylation (unscaled β-values). Colored horizontal labels indicate IGHV mutation status (U- or M-CLL) and BR (low or high BR, at the threshold of 0.35% cells/day). (B) PCA of DNA methylation data from all fractions, colored by either IGHV mutational status or BR. (C) Scatter plots for pairwise comparisons of DNA methylation between U-CLL and M-CLL (top) or between low BR and high BR (bottom) patients by CXCR4/CD5 intraclonal fractions. Sites were highlighted at false discovery rate (FDR) <0.05 (black dots), and further considered significant at FDR <0.01 and absolute mean methylation difference of ≥10% (red dots). Colors in the cartoon representation of cell fractions are maintained throughout the panels. (D) Venn diagram showing relations between common overlap (c-DMC) and fraction-specific differentially methylated CpGs (f-DMC), which were identified as red dots in panel C after pairwise comparison of U- and M-CLL patients. (E) Heatmap of c-DMCs and f-DMCs between U- and M-CLL, ordered by IGHV status and by fractions. Colored horizontal labels indicate other established predictors of CLL progression: CD38+ (>30% positive), ZAP70+ (>20% positive). Framed quadrants in the heatmap highlights f-DMCs initially identified in the corresponding fraction, despite largely comparable methylation profiles. IF, intermediate fraction; PC, principal component; PF, proliferative fraction; RF, resting fraction.
Intraclonally stable methylation patterns in CLL reflect signatures of the potential normal B-cell of origin. (A) Unsupervised HC of the fractions by Manhattan distance measurement of DNA methylation (unscaled β-values). Colored horizontal labels indicate IGHV mutation status (U- or M-CLL) and BR (low or high BR, at the threshold of 0.35% cells/day). (B) PCA of DNA methylation data from all fractions, colored by either IGHV mutational status or BR. (C) Scatter plots for pairwise comparisons of DNA methylation between U-CLL and M-CLL (top) or between low BR and high BR (bottom) patients by CXCR4/CD5 intraclonal fractions. Sites were highlighted at false discovery rate (FDR) <0.05 (black dots), and further considered significant at FDR <0.01 and absolute mean methylation difference of ≥10% (red dots). Colors in the cartoon representation of cell fractions are maintained throughout the panels. (D) Venn diagram showing relations between common overlap (c-DMC) and fraction-specific differentially methylated CpGs (f-DMC), which were identified as red dots in panel C after pairwise comparison of U- and M-CLL patients. (E) Heatmap of c-DMCs and f-DMCs between U- and M-CLL, ordered by IGHV status and by fractions. Colored horizontal labels indicate other established predictors of CLL progression: CD38+ (>30% positive), ZAP70+ (>20% positive). Framed quadrants in the heatmap highlights f-DMCs initially identified in the corresponding fraction, despite largely comparable methylation profiles. IF, intermediate fraction; PC, principal component; PF, proliferative fraction; RF, resting fraction.
Next, we explored DNA methylation differences between U-CLL and M-CLL samples in the individual sorted fractions and identified a large number of differentially methylated CpGs (DMCs) between the 2 prognostic groups in each of the fractions (Figure 2C). Venn diagram comparisons demonstrated that 9966 of these DMCs were common to all 4 fractions (c-DMCs) (Figure 2D; supplemental Table 1). Notably, 2763 DMCs initially appeared to be fraction specific (f-DMCs) at the given statistical cutoff (941 in PF, 291 in IF, and 1531 in RF) and were not detected in unseparated tumor cells because of the underlying heterogeneity (Figure 2D; supplemental Table 2). However, these f-DMCs exhibited identical directionality in methylation differences in all intraclonal fractions (Figure 2E). In fact, IGHV mutation–associated c-DMCs and f-DMCs similarly included both hypo- and hypermethylated sites that were significantly underrepresented in CpG islands and enriched in “open sea” regions (supplemental Figure 1B). Using functional chromatin segmentation,34 we observed that M-CLL displayed hypomethylation of enhancers and weak transcriptional regulation sites, while hypermethylation occurred in some enhancers but was most evident in heterochromatic regions (supplemental Figure 1C). These results confirm previous established associations of DNA methylation with the IGHV mutational status in CLL20,25 and expand on those findings by demonstrating that such methylation patterns are stably maintained across the circulating fractions. However, as will be confirmed below, these cells do appear to undergo subtle epigenetic changes as they move further away from the time of their last cell division.
Established intraclonal methylation patterns reflect those of late B-cell maturation stages
Next, we sought to compare the epigenetic state of our CLL samples with published methylomes from normal human primary B-cell subtypes.26 Supplemental Figure 1D shows a phylogenetic tree based on Manhattan distances between the individual patients based on the total fractions. Consistent with the highly conserved methylation state across the CXCR4/CD5 fractions and the bulk CLL samples, similar results were obtained using the fractions (not shown). A common maturation axis suggested a continuum of DNA methylation, where M-CLL samples exhibited methylation states closer to those of the more differentiated memory B cells or bone marrow plasma cells, while U-CLL samples clustered between naive and germinal center (GC) or early-post-GC stages (tonsil plasma cells), which was suggestive of an early-GC epigenetic hallmark. This was consistent with previous postulations that the IGHV-related DNA methylation pattern likely derives from the founder cell and that U-CLL cells are representative of a less mature B-cell differentiation stage than M-CLL cells.20,25,31,35
Further supporting this notion, a set of 5 previously described CpG biomarkers that can be used to classify CLL methylomes into 3 epigenetic subgroups related to normal counterpart B-cell differentiation stages24 was found among the c-DMCs identified here (supplemental Table 1) and suggested that U-CLL cells bear a methylation phenotype closer to naive B cells than M-CLL cells (Table 1). In addition, this classifier successfully predicted outcome in our population (supplemental Figure 1E). Interestingly, even though patient CLL1039 was classified as M-CLL, its epigenetic profile was more similar to the U-CLL subgroup (supplemental Figure 1D), and this was more consistent with its poor clinical outcome, reflected in a short TTFT, high BR, and chromosomal abnormalities (Table 1). Indeed, the 5-CpG algorithm24 classified CLL1039 as an intermediate case, confirming that the epigenetic cell-of-origin categorization can complement the prognostic value of IGHV mutation status, BR, and other well-known factors, especially for the M-CLL subgroup.
BRs show limited impact on DNA methylation of circulating CLL fractions
Since plasticity and changes of the DNA methylome (mainly hypomethylation) accompany B-cell proliferation,25,26,35 we next investigated whether different leukemia BRs associated with changes in DNA methylation. As discussed above, unsupervised HC and PCA analysis failed to reveal a distinctive global association of DNA methylation and BR in any of the cellular fractions (Figure 2A-B), mainly because of the predominant differences between U- and M-CLL samples. Indeed, a marked overlap of IGHV mutation status and BR can be observed (Figure 1B and 2A), complicating the identification of an independent impact of proliferation rates on the CLL methylome. However, results from the larger clinical trial to which this study is linked demonstrated that increased BR of CLL cells holds an independent prognostic value that can be added to the well-accepted prognostic value of IGHV status.28 To expand on this finding, we compared the DNA methylation landscape of fractions from patients with high and low BR, correcting for IGHV status, and found a low number of loci, especially in the RF, that were significantly differentially methylated, more often showing hypermethylation associated with higher BR (Figure 2C; supplemental Table 3). These results suggest that beyond the baseline epigenetic impact associated to IGHV mutation status, BR has a limited impact on CLL DNA methylation levels, which is ultimately obscured in the bulk clone.
Increased DNA methylation heterogeneity further accumulates in resting CXCR4BrightCD5Dim CD19+ cells
Active transition of B cells through the GC may also be revealed by the accumulation of DNA methylation heterogeneity.36-38 To explore whether U-CLL and M-CLL fractions accumulated different levels of DNA methylation heterogeneity as a consequence of a distinct cell-of-origin history, we evaluated interpatient heterogeneity through calculation of pairwise Manhattan distances of their mean methylation levels (Figure 3A) and their correlation coefficients (Figure 3B). We observed that all fractions from M-CLL patients exhibited a significantly increased interpatient methylation heterogeneity compared with U-CLL and, correspondingly, poorer pairwise correlations. Such variegation in DNA methylation is consistent with the proposed GC/early-post-GC origin of the M-CLL subtype20 (supplemental Figure 1D).
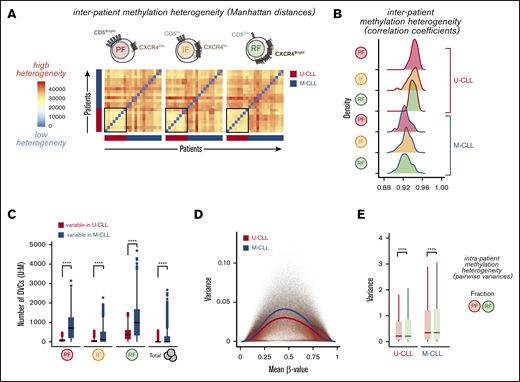
Interpatient and intraclonal DNA methylation heterogeneity. (A) Heatmaps showing pairwise methylation Manhattan distances indicating the level of heterogeneity within each leukemic fraction among all patients. Vertical and horizontal bars indicate the IGHV status of the individual patients. (B) Density histograms of pairwise Pearson correlation coefficients within each leukemic fraction among all patients, separated by IGHV mutational status. Significant differences were observed between U-CLL and M-CLL distributions of correlation coefficients in all fractions (P ≤ 5.174 × 10e-7, Wilcoxon rank sum test with continuity correction). (C) Box plot showing the number of differentially variable CpG sites (DVCs, by DiffVar method) in each of the fractions with significant differences in group variances between U-CLL and M-CLL patients, determined by using all possible permutations of equal number of samples per group (n = 8). (D) Plot of variance and mean methylation levels (β-values) for each CpG site across U-CLL or M-CLL samples. Color lines represent average variance in each subgroup illustrating the higher variance at intermediate β-values and in the M-CLL samples. (E) Box plot showing the distribution of variances for every CpG in the resting (RF) and proliferative (PF) intraclonal fractions, separated by U-CLL (n = 8) and M-CLL (n = 13) classification. Both the Fligner-Killeen test of homogeneity of variances and the paired Wilcoxon signed-rank test were used to compare CpG methylation variances with similar significant results between the paired fractions. ****P < .0001.
Interpatient and intraclonal DNA methylation heterogeneity. (A) Heatmaps showing pairwise methylation Manhattan distances indicating the level of heterogeneity within each leukemic fraction among all patients. Vertical and horizontal bars indicate the IGHV status of the individual patients. (B) Density histograms of pairwise Pearson correlation coefficients within each leukemic fraction among all patients, separated by IGHV mutational status. Significant differences were observed between U-CLL and M-CLL distributions of correlation coefficients in all fractions (P ≤ 5.174 × 10e-7, Wilcoxon rank sum test with continuity correction). (C) Box plot showing the number of differentially variable CpG sites (DVCs, by DiffVar method) in each of the fractions with significant differences in group variances between U-CLL and M-CLL patients, determined by using all possible permutations of equal number of samples per group (n = 8). (D) Plot of variance and mean methylation levels (β-values) for each CpG site across U-CLL or M-CLL samples. Color lines represent average variance in each subgroup illustrating the higher variance at intermediate β-values and in the M-CLL samples. (E) Box plot showing the distribution of variances for every CpG in the resting (RF) and proliferative (PF) intraclonal fractions, separated by U-CLL (n = 8) and M-CLL (n = 13) classification. Both the Fligner-Killeen test of homogeneity of variances and the paired Wilcoxon signed-rank test were used to compare CpG methylation variances with similar significant results between the paired fractions. ****P < .0001.
To further identify differentially variable CpG sites (DVCs) in both U- and M-CLL groups, where the fractions in one group have consistent methylation values and the fractions in the other group have highly variable methylation values, we applied the DiffVar method39 (see supplemental Methods) to our data. We observed a significant increase in the number of variable loci in all M-CLL fractions (Figure 3C), again supporting the intrinsically higher heterogeneity of M-CLL patients compared with U-CLL. Indeed, despite the expected effect of stochastic loss of DNA methylation and increased variance at partially methylated sites (Figure 3D), we still could observe a higher variance in M-CLL patients across the full range of β-values. In both patient subtypes, however, this variability seemed especially evident within the longer-lived CLL cells of the RF, but less pronounced in the recently born CLL cells of the PF (Figure 3C). Consistent with this, both the distributions of variances and the Fligner-Killeen test of homogeneity of variances between PF and RF for each CpG (pairwise) showed a significantly higher variance in the RF in both U- and M-CLL groups (Figure 3E), suggesting that DNA methylation heterogeneity further accumulates as cells age in the circulation without dividing.
Longevity-linked DNA methylation changes accumulate in recently born CXCR4DimCD5Bright CD19+ cells
The evidence for “time since last division” as a potential source of intraclonal heterogeneity in both U-CLL and M-CLL patients prompted us to investigate the nature of these methylation changes and whether they might occur recurrently at specific loci. To explore this, we focused on intraclonal differences in CpG methylation, rather than interpatient variability, by using paired analysis that compared fractions within each patient (Figure 4). M-CLL and U-CLL subgroups were analyzed separately to investigate whether their apparently different cell-of-origin and IGHV status might also associate with distinct epigenetic instability as cells age. U-CLL patients evidenced 533 loci that recurrently accumulated significant epigenetic differences when comparing PF and RF intraclonal fractions (Figure 4A). However, these 533 DMCs identified in U-CLL cells (Figure 4B) showed the same tendency of methylation change in the M-CLL cells (Figure 4C; supplemental Table 4), which may have been obscured by the intrinsic heterogeneity of M-CLL (Figure 3) and is consistent with a common linear epigenetic advancement in the circulating fractions of both U-CLL and M-CLL patients.
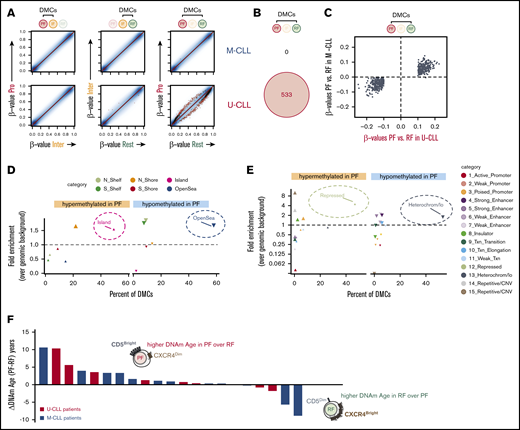
CLL fractions revealed an intraclonal program of accumulated epigenetic changes linked to the longevity of the cells. (A) Scatter plots for pairwise comparisons of DNA methylation between fractions. Differentially methylated CpGs are highlighted as black (FDR-adjusted P < .05) or red (FDR-adjusted P < .05 and β-value difference ≥10%) dots, based on paired linear models generated with the R/Bioconductor package limma. (B) Venn diagram showing IGHV-specific differential methylation between PF and RF (red dots in panel A). (C) Scatter plot illustrating similar tendencies in U-CLL (x-axis) and M-CLL (y-axis) patients for those differences in DNA methylation β-values primarily identified only in U-CLL between RF and PF (red dots in panel A). Relative distribution of DMCs between PF and RF in relation to CpG islands (D) or across chromatin states identified in the lymphoblastoid cell line GM12878 by ChromHMM (E). Distribution of hypomethylated sites is depicted by downward-pointing triangles, while distribution of hypermethylated sites is depicted by upward-pointing triangles. The y-axis shows fold enrichment for the corresponding DMCs over the frequency of all measured CpGs within the same regions, and the x-axis shows the percent of the indicated DMCs that occur in each annotation feature depicted by different colors. Larger symbol sizes depict P < .01. (F) The rate of methylation aging during the CLL life cycle is displayed as difference in DNAm age (years) between the PF and RF intraclonal fractions. Individual CLL patients are color-coded by IGHV mutation status and ranked by absolute difference in the recently born PF cells.
CLL fractions revealed an intraclonal program of accumulated epigenetic changes linked to the longevity of the cells. (A) Scatter plots for pairwise comparisons of DNA methylation between fractions. Differentially methylated CpGs are highlighted as black (FDR-adjusted P < .05) or red (FDR-adjusted P < .05 and β-value difference ≥10%) dots, based on paired linear models generated with the R/Bioconductor package limma. (B) Venn diagram showing IGHV-specific differential methylation between PF and RF (red dots in panel A). (C) Scatter plot illustrating similar tendencies in U-CLL (x-axis) and M-CLL (y-axis) patients for those differences in DNA methylation β-values primarily identified only in U-CLL between RF and PF (red dots in panel A). Relative distribution of DMCs between PF and RF in relation to CpG islands (D) or across chromatin states identified in the lymphoblastoid cell line GM12878 by ChromHMM (E). Distribution of hypomethylated sites is depicted by downward-pointing triangles, while distribution of hypermethylated sites is depicted by upward-pointing triangles. The y-axis shows fold enrichment for the corresponding DMCs over the frequency of all measured CpGs within the same regions, and the x-axis shows the percent of the indicated DMCs that occur in each annotation feature depicted by different colors. Larger symbol sizes depict P < .01. (F) The rate of methylation aging during the CLL life cycle is displayed as difference in DNAm age (years) between the PF and RF intraclonal fractions. Individual CLL patients are color-coded by IGHV mutation status and ranked by absolute difference in the recently born PF cells.
In the PF, most of these intraclonal DMCs accumulated hypomethylation in open sea and heterochromatic regions, while hypermethylation was mainly enriched in CpG islands and Polycomb-repressed regions (Figure 4D-E). Interestingly, this signature has been previously linked to aging, extended cellular lifespan, and cancer and has recently been attributed to normal B-cell differentiation and ongoing GC-like experience.26,35 Furthermore, when we used a previously described epigenetic clock based on DNA methylation biomarkers of aging (DNAm Age)40 as a surrogate marker for contrasting the ages of PF and RF in each patient, we observed that in 80% of cases (17 out of 21), the estimated DNAm Age of the PF was higher or at least comparable to that of the RF (Figure 4F). Altogether, these results reveal that intraclonal CLL fractions in U-CLL and, to a lesser extent, M-CLL patients exhibit subtle epigenetic changes as they advance in a progressive life cycle of CLL, with recently born PF cells being more ancient and carrying acquired longevity-linked DNA methylation changes.
Intraclonal differential expression confirms the proliferative histories of the fractions and reveals unique associations with clinical outcome
Despite the poor overall correlation between gene expression and IGHV mutation status or BR (supplemental Figure 2), we expected to confirm our previous observations that the fractions reveal expression differences related to their proliferative histories.6 Indeed, we found 448 differentially expressed genes (DEGs) between paired PF and RF fractions (supplemental Table 5), which intersected with and followed a similar directionality with those originally reported6 (Figure 5A). As expected, messenger RNA levels for CXCR4 and CD5 were consistent with the protein expression of these receptors used for performing the FACS-based fractionation (Figure 5B).
![Differentially expressed genes between CLL fractions show unique features associated with cell survival and patient outcome. (A) Venn diagrams showing overlaps between our set of DEGs in RF vs PF cells and those previously described.6 Those overlaps were significantly enriched as measured by Fisher's exact tests (combined DEGs: P < 5e-320, odds ratio [OR] = 61.9; DEGs up in PF: P < 2e-268, OR = 90.1; DEGs up in RF: P = 1e-112, OR = 64.8). (B) Comparative expression of the 2 genes CXCR4 and CD5 that inversely change their levels and were used for FACS sorting of PF and RF. CXCR4 was significantly upregulated in RF (P < 5e-8 in U-CLL and P < 3e-6 in M-CLL), while CD5 was significantly more expressed in PF (P < 9e-6 in U-CLL and P < 5e-7 in M-CLL). (C) Venn diagram showing overlaps of intraclonal DEGs that associate with TTFT uniquely in either PF or RF as identified by Cox proportional hazards model or by log-rank test after stratifying patients according to gene expression levels. (D) Scatter plots of log2 fold-change vs log10P values in differential expression analysis from PF to RF (dots represent Illumina probes). Only DEGs associated with TTFT in PF (top) or RF (bottom) and overlapping in panel C are shown. (E) Comparative expression of representative intraclonal DEGs associated with TTFT and either upregulated in PF (CCND2, ILMN_2067656) or upregulated in RF (TJAP1, ILMN_1743763). (F) Kaplan-Meier curves of TTFT for CLL patients stratified according to an optimal cutoff for CCND2 (ILMN_2067656) or TJAP1 (ILMN_1743763) expression levels. P values were determined using the log-rank test. TTFT is in months.*P < .05, **P < .01, ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/5/10.1182_bloodadvances.2019000817/3/m_advancesadv2019000817f5.png?Expires=1765918722&Signature=USaV-ZyZk6u7W3ZOw6BbUBYh~5hbTAirOrJsIJynv1ix5fvvMVBeBo88m2csaw~9rJ2-A~T8OY8klEX-FMZ2h9sFNna-OS4OVTgJ1GQdIGc5gQBxRSn8xFOg7S66Jw3Gz5n4c1CyU29GID7op1NwyFYqSdx9QKTkY88DCrklJGxQaf236eGkP1kLLYI26sMsZ629HdXyhOnoECi7kgm21OFKLX3ajqvLZrUXO8qZfhPYeKsCxi-JMu-08zUwC8qwVKTdBIrG9NapB58Tin0Bfz9XuXbXybL0iSQD2THAbOinuIzrixYBtRjkEypVXoA96xshD6kphsESMqrNjDUS8Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Differentially expressed genes between CLL fractions show unique features associated with cell survival and patient outcome. (A) Venn diagrams showing overlaps between our set of DEGs in RF vs PF cells and those previously described.6 Those overlaps were significantly enriched as measured by Fisher's exact tests (combined DEGs: P < 5e-320, odds ratio [OR] = 61.9; DEGs up in PF: P < 2e-268, OR = 90.1; DEGs up in RF: P = 1e-112, OR = 64.8). (B) Comparative expression of the 2 genes CXCR4 and CD5 that inversely change their levels and were used for FACS sorting of PF and RF. CXCR4 was significantly upregulated in RF (P < 5e-8 in U-CLL and P < 3e-6 in M-CLL), while CD5 was significantly more expressed in PF (P < 9e-6 in U-CLL and P < 5e-7 in M-CLL). (C) Venn diagram showing overlaps of intraclonal DEGs that associate with TTFT uniquely in either PF or RF as identified by Cox proportional hazards model or by log-rank test after stratifying patients according to gene expression levels. (D) Scatter plots of log2 fold-change vs log10P values in differential expression analysis from PF to RF (dots represent Illumina probes). Only DEGs associated with TTFT in PF (top) or RF (bottom) and overlapping in panel C are shown. (E) Comparative expression of representative intraclonal DEGs associated with TTFT and either upregulated in PF (CCND2, ILMN_2067656) or upregulated in RF (TJAP1, ILMN_1743763). (F) Kaplan-Meier curves of TTFT for CLL patients stratified according to an optimal cutoff for CCND2 (ILMN_2067656) or TJAP1 (ILMN_1743763) expression levels. P values were determined using the log-rank test. TTFT is in months.*P < .05, **P < .01, ***P < .001.
Differentially expressed genes between CLL fractions show unique features associated with cell survival and patient outcome. (A) Venn diagrams showing overlaps between our set of DEGs in RF vs PF cells and those previously described.6 Those overlaps were significantly enriched as measured by Fisher's exact tests (combined DEGs: P < 5e-320, odds ratio [OR] = 61.9; DEGs up in PF: P < 2e-268, OR = 90.1; DEGs up in RF: P = 1e-112, OR = 64.8). (B) Comparative expression of the 2 genes CXCR4 and CD5 that inversely change their levels and were used for FACS sorting of PF and RF. CXCR4 was significantly upregulated in RF (P < 5e-8 in U-CLL and P < 3e-6 in M-CLL), while CD5 was significantly more expressed in PF (P < 9e-6 in U-CLL and P < 5e-7 in M-CLL). (C) Venn diagram showing overlaps of intraclonal DEGs that associate with TTFT uniquely in either PF or RF as identified by Cox proportional hazards model or by log-rank test after stratifying patients according to gene expression levels. (D) Scatter plots of log2 fold-change vs log10P values in differential expression analysis from PF to RF (dots represent Illumina probes). Only DEGs associated with TTFT in PF (top) or RF (bottom) and overlapping in panel C are shown. (E) Comparative expression of representative intraclonal DEGs associated with TTFT and either upregulated in PF (CCND2, ILMN_2067656) or upregulated in RF (TJAP1, ILMN_1743763). (F) Kaplan-Meier curves of TTFT for CLL patients stratified according to an optimal cutoff for CCND2 (ILMN_2067656) or TJAP1 (ILMN_1743763) expression levels. P values were determined using the log-rank test. TTFT is in months.*P < .05, **P < .01, ***P < .001.
To investigate whether specific expression patterns in the fractions might be more predictive of clinical outcome than looking at the bulk cells, we used Cox regression and log-rank analyses to identify intraclonal DEGs that preserved their prognostic potential in both analyses (Figure 5C; supplemental Table 6). Some exhibited more than twofold difference in expression between PF and RF (Figure 5D). As examples, high levels of cyclin D2 in the PF or of tight junction associated protein 1 (TJAP1) in the RF (Figure 5E), were able to dichotomize clinical outcome of the 21 CLL patients only when the corresponding fraction was interrogated (Figure 5F). Consistent with this, it has been shown that cyclin D2 is critical for CD5+ B cells41 and is overexpressed in proliferation centers of CLL,42 while TJAP1 has been observed in B-CLL cell lines.43 Together, these results suggest that transcriptional regulation of some genes in the fractions, especially in the PF, might have a significant impact in the overall clinical progression of the disease that would not be appreciated looking at the bulk CLL clone.
Discussion
The majority of leukemic cells in CLL reside in peripheral lymphoid organs, where almost all tumor cell division occurs.44 However, a constant egress from solid tissues generates easily accessible circulating CD19+CD5+ tumor cells routinely collected to study patient prognosis and disease etiology and pathogenesis.1,2 We hypothesized that the analysis of intraclonal cellular subpopulations and the transitions between them would reveal new prognostic markers, patterns of pathogenesis, or insights into the cell of origin of the tumor, which might have been obscured previously by the heterogeneity of the total circulating population. Here, we used the reciprocal expression of CXCR4 and CD5 surface markers on circulating tumor cells from newly diagnosed untreated CLL patients to obtain fractions enriched for cells that had recently divided, presumably in the tissues, and older cells that were quiescent, having divided earlier.
Unsupervised HC of the fractions and of the bulk tumor cells based on DNA methylation separated samples consistent with their IGHV mutation status. Many of the observed methylation differences between U-CLL and M-CLL were shared by all intraclonal fractions and had identical directionality, which in combination with other evidence31,45 supports the existence of a remarkably stable overall pattern of DNA methylation in CLL. This most often consisted of hypomethylation at “open sea” CpG sites in enhancers and weakly transcribed regions in M-CLL patients compared with U-CLL. Interestingly, this hallmark had been previously observed in CLL20,25 and had been compared with the loss of methylation that characterizes normal B-cell development,25,26,38 suggesting a more mature cell of origin for M-CLL than U-CLL patients. Furthermore, evaluation of interpatient methylation heterogeneity revealed a slightly increased variability and poorer pairwise correlations in fractions obtained from M-CLL patients compared with U-CLL. Such increased variability resembles the accumulation of DNA methylation heterogeneity observed during normal transition through the GC36,37,46 and further supports the pre- or early-GC cell of origin suggested for U-CLL cells. Indeed, our phylogenetic analysis comparing CLL fractions to normal B cells also positioned M-CLL methylomes closer to memory or bone marrow plasma cells, while U-CLL methylomes suggested a slightly less differentiated state. The fact that these differences in methylation profiles were found in all fractions of U-CLL and M-CLL patients and included the 5 previously described CpG biomarkers with the potential to assign a normal B-cell counterpart to the leukemic cell24 further supports the idea that the dominant methylation hallmark reflects the cell of origin. It is important to note that these interpatient epigenetic differences were not clearly associated with the BR determined by heavy water labeling, even in the PF that had recently undergone cell division. This could be either because DNA methylation is independent of BR or because any potential proliferation-related epigenetic signature disappears very soon after leukemic cells leave the proliferation centers and move into the circulation. Analogously, normal naive B cells from tonsils or peripheral blood exhibit comparable DNA methylation profiles, even though they have significantly different transcriptomes.26 On the basis of these observations, we conclude that the rate of proliferation and physical egression out of the lymphoid niche into the bloodstream have a limited impact on the methylome of the CLL cell.
Paired analysis of differentially methylated CpGs between fractions from each of the U-CLL and M-CLL patients revealed that in both subtypes, but more clearly in the U-CLL group, the cells of the PF differed from those of the RF based on hypomethylation in open-sea and heterochromatic regions and hypermethylation mainly in CpG islands and Polycomb-repressed regions. Interestingly, this signature has been previously linked not only to solid and hematological tumors but also to aging, extended cellular lifespan, and normal B-cell differentiation.26,47 As cells in the RF are thought to derive from the PF after time in circulation without dividing, this represents a paradox that PF methylome is “older” than RF. Especially since we observed that long-lived RF cells carried more CpG sites with heterogeneous methylation than recently born (or reborn) cells of the PF, and such acquired heterogeneity might become a potential source for additional epigenetic aging and genetic evolution as time passes in circulation since the last division. One possibility to reconcile this is in a CLL life cycle where PF cells may be linear descendants, therefore inheriting the epigenetic age, of some resting cells that reentered proliferation centers and further acquired aging-associated epigenetic changes upon cell division (see model in Figure 1A and visual abstract). In every life cycle of the leukemic cells, it might be expected that any RF-related epigenetic signature would either be reset as leukemic cells pass through the proliferation centers, or stay and accumulate in every round of recently born PF. The former seems less likely, since the dominant IGHV mutation–related epigenetic signature remains quite stable, supporting the pre-existence and retention of these epigenetic states in all fractions as constitutive cell-of-origin hallmarks that are not acquired or reshaped during the life cycle of the leukemic cell. Cumulative hallmarks, on the other hand, become hard to identify without comparison of sequential samples from the same patients during progression of the disease. However, our evidence suggests a progressive epigenetic aging of the newly proliferated PF, consistent with cells of the PF being linear descendants of some cells from the RF that reentered lymphoid solid tissue and received prosurvival stimuli to proliferate again. However, based on the available data, we cannot rule out the possibility that the PF derives from a distinct set of cells, independent of the RF, that continuously undergo rounds of replication. In this nonexclusive scenario, DNAm Age may be increasing at an accelerated rate in the proliferation centers/lymphoid niche due to the stimulation and proliferation of CLL cells, and once released from this environment, the mAge of CLL cells advances at a slower rate (in most patients). Thus, RF cells at a given sampling time point would have been produced from PF cells that were released from the proliferation centers at an earlier date and thus exposed to the mAge-accelerating environment for less time. PF cells present in the sample would have been released at a later date thus exposed to the DNAm Age–accelerating environment for more time, contributing to PF being epigenetically “older” than RF in most patients. While future research is still required to solve these questions, our observations are consistent with whole-genome sequencing of indolent CLL that has recently revealed an ongoing ageing-related signature based on spontaneous deamination of CpG sites that results in genetic mutations.48,49
Largely independent of DNA methylation, CLL fractions revealed intraclonal gene expression differences that confirmed the expected variation of CXCR4/CD5 surface levels and supported the advancement of the leukemic cell from the proliferative to the resting compartment6 in a clonally related continuum of CD5 states.50 Importantly, a substantial number of differentially expressed genes in the PF associated with TTFT in the patients, demonstrating a clinical prognostic potential that was obscured in the bulk circulating clone and suggesting that the initial transcriptional profile in this discrete compartment might have consequences in disease progression. Further studies are warranted to investigate the biological significance of these PF-associated genes.
In summary, by profiling different CXCR4/CD5 circulating CLL intraclonal fractions, we were able to identify both pre-established and more recently acquired molecular hallmarks. While persistent DNA methylation is suggestive of late mature B-cell origins, more subtle acquired DNA methylation patterns support a quiescent aging of the leukemic cells while advancing from the PF to the RF and then back to the PF after passing through proliferation centers. Thus, the linearity of our proposed CLL life cycle (Figure 1A) is supported (1) by the transcriptional differences that have been observed previously6 and here between fractions, which affect survival and cell movement pathways and reveal clinical associations with an earlier TTFT that could not be detected in bulk cells; and (2) by the observation that the PF seems epigenetically more homogeneous, with additional aging-related epigenetic changes acquired either as the cell divides just before entering the circulation or in circulation and that might then be carried back into the next round of reborn PF cells. These findings highlight the importance of further research and exploration of therapeutic strategies for targeting the PF while accelerating the senescence and death of the RF to block the reentrance of CLL resting cells into a new life cycle.
The data reported in this article have been deposited in the Gene Expression Omnibus database (SuperSeries accession number GSE144901).
Acknowledgments
This work was supported in part by a Lymphoma Research Foundation CLL/SLL collaborative grant (N.C. and M.D.S.) and National Institutes of Health, National Cancer Institute grants R01-CA081554 (N.C.) and 5R01CA072649 (M.D.S.) and National Institute of Allergy and Infectious Diseases grants 5R01AI132507 and 9R01AI112335 (M.D.S.) S.R. was funded by Ministerio de Economía y Competitividad (SAF2013-45787-R and SAF2017-82309-R) cofunded by European Regional Development Fund, the Ramón y Cajal program (RYC-2014-16399) cofunded by European Social Fund, Gobierno de Navarra (GN-106/2014), and contributions from Fundación La Caixa. The authors acknowledge John Byrd, Jeffrey Jones, and Thomas Lin from The Ohio State University for contributing samples for analysis; these samples were collected during the mentioned study by Murphy et al28 and were cryopreserved and stored in the Tissue Core (Laura Rassenti, head) of the National Institutes of Health, National Cancer Institute–funded PO1 program “CLL Research Consortium” (Thomas J. Kipps, principal investigator). The authors also acknowledge philanthropic contributions from The Nash Family Foundation, The Marks Foundation, the Karches Family, the Mona and Edward Albert Foundation, and the Jean Walton Fund for Leukemia, Lymphoma, and Myeloma Research.
Authorship
Contribution: B.A.B. and S.R. performed computational studies and analyzed and interpreted the data; X.W., X.-J.Y., N.C., M.D.S., and S.R. designed the research; X.W., X.-J.Y., M.P., and M.F. performed experiments; J.B., S.L.A., J.A.M.-C., and K.R.R. discussed the results and revised the manuscript; N.C., M.D.S., X.W., and S.R. conceived the study; and N.C., M.D.S., B.A.B., and S.R. cowrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Matthew D. Scharff, Albert Einstein College of Medicine, 1300 Morris Park Ave, Bronx, NY 10461; e-mail: matthew.scharff@einsteinmed.org; Nicholas Chiorazzi, The Feinstein Institute for Medical Research, 350 Community Dr, Manhasset, NY 11030; e-mail: nchizzi@northwell.edu; and Sergio Roa, Cima Universidad de Navarra, 55 Av Pio XII, 31008 Pamplona, Navarra, Spain; e-mail: sroa@unav.es.

