

Key Points
CD300f is expressed in the majority of AML patients and across HSPCs; an anti-CD300f ADC depletes AML cells and HSPCs.
In vitro and in vivo data suggest targeted CD300f therapy enhances efficacy and reduces toxicity of HSCT in AML.

Abstract
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) significantly reduces the rate of relapse in acute myeloid leukemia (AML) but comes at the cost of significant treatment-related mortality. Despite the reduction in relapse overall, it remains common, especially in high-risk groups. The outcomes for patients who relapse after transplant remains very poor. A large proportion of the morbidity that prevents most patients from accessing allo-HSCT is due to toxic nonspecific conditioning agents that are required to remove recipient hematopoietic stem and progenitor cells (HSPCs), allowing for successful donor engraftment. CD300f is expressed evenly across HSPC subtypes. CD300f has transcription and protein expression equivalent to CD33 on AML. We have developed an anti-CD300f antibody that efficiently internalizes into target cells. We have generated a highly potent anti-CD300f antibody-drug conjugate (ADC) with a pyrrolobenzodiazepine warhead that selectively depletes AML cell lines and colony forming units in vitro. The ADC synergizes with fludarabine, making it a natural combination to use in a minimal toxicity conditioning regimen. Our ADC prolongs the survival of mice engrafted with human cell lines and depletes primary human AML engrafted with a single injection. In a humanized mouse model, a single injection of the ADC depletes CD34+ HSPCs and CD34+CD38−CD90+ hematopoietic stem cells. This work establishes an anti-CD300f ADC as an attractive potential therapeutic that, if validated in transplant models using a larger cohort of primary AML samples, will reduce relapse rate and toxicity for patients with AML undergoing allo-HSCT.
Introduction
Relapse after allogeneic hematopoietic stem cell transplant (allo-HSCT) for acute myeloid leukemia (AML) occurs in 24% to 36% of patients, and the outcomes for these patients are poor.1 Disease genetic characteristics can predict for relapse overall and impact post–allo-HSCT relapse rates.2 The rate of relapse after allo-HSCT is higher in adverse-risk groups, particularly in some subgroups such as monosomal karyotypes.3,4 Postinduction factors that predict relapse include the presence of residual disease. Minimal residual disease (MRD) positivity prior to allo-HSCT, detected by flow cytometry, quantitative polymerase chain reaction, or next-generation sequencing, correlates with relapse.5-7 Although allo-HSCT remains the only potential curative option in patients with refractory disease, relapse rates remain high in that setting.8
The role of the immune response and graft-versus-leukemia effect is well established.9 Evidence demonstrates that the intensity of conditioning plays a clear role in reducing relapse risk. Myeloablative (MA) allo-HSCT conditioning regimens reduce relapse more than reduced-intensity conditioning (RIC) and non-MA regimens.10 The increased relapse rate seen in patients who are MRD positive or undergo non-MA conditioning suggests that reducing the burden of disease by the time of transplant is critical to improving outcomes.
The advent of RIC and non-MA regimens has transformed transplantation, making it accessible to older patients and those with comorbidities. RIC regimens demonstrate significantly less treatment-related mortality (TRM) than MA regimens.11 Despite the reduction seen in RIC, TRM remains significant, especially in those >65 years.12 The development of antibody-based therapies depleting hematopoietic stem and progenitor cells (HSPCs) as part of allo-HSCT conditioning is expanding.13 Such therapies may reduce or eliminate traditional methods of depleting HSPC such as alkylating agents and irradiation. Preclinical studies demonstrate that antibody-drug conjugate (ADC)–based conditioning limits damage to bone marrow (BM) architecture and accelerates immune recovery compared with traditional conditioning.14 The advent of targeted condition has the potential to further reduce TRM.
The CD300f protein (encoded by the CD300LF gene) is an inhibitory receptor found on healthy myeloid cells, including antigen-presenting cells (APCs).15,16 CD300f is present on a high proportion of AML cells as well as HSPCs.17,18 Its distribution makes CD300f an excellent target in both AML therapy and targeted allo-HSCT conditioning. We have completed proof-of-principle work demonstrating how incorporating an anti-CD300f ADC into conditioning for allo-HSCT in AML may decrease relapse and toxicity by reducing/replacing traditional agents.
Methods
Preparation of tissue samples
Blood and BM samples from patients with AML or healthy individuals were collected at Concord Repatriation General Hospital (CRGH) or Royal Prince Alfred Hospital (Sydney, Australia). Patient and sample demographics are provided in supplemental Table 1. Peripheral blood (PB) or BM samples from healthy donors were collected at CRGH. Cord blood (CB) samples were collected by the Sydney CB Bank. Donors provided informed consent under ethical approval obtained from the Sydney Local District Human Research Ethics Committee (HREC/12/CRGH/59, HREC/11/CRGH/61 and 118). Mononuclear cells (MCs) were isolated from samples using density gradient centrifugation through Ficoll-Paque Plus (GE Healthcare) according to the manufacturer’s protocols. Samples were passed through a 22G needle to disrupt BM fragments and then filtered prior to MC isolation as above. Human monocytes were purified from MCs by CD14 MicroBeads selection using an AutoMACS Pro (Miltenyi Biotec).
Cell lines
The AML cell lines HL-60 and THP-1 were obtained by Derek Hart at the Christchurch School of Medicine, University of Otago. The U973, Z-138, and Mino cell lines were from the American Type Culture Collection. All cell lines were maintained in complete RPMI 1640 (complete RPMI) containing 10% fetal calf serum, 2 mM Gluta-MAX, 100 U/mL penicillin, and 100 μg/mL streptomycin (ThermoFisher).
Gene expression
AML gene expression data were retrieved from the Gene Expression Omnibus microarray dataset GSE14468.19 HSPC gene expression was retrieved from GSE42519, GSE17054, and GSE19599.20-22 The series matrix files were parsed in R, and the probe ID and signal values corresponding to CD300LF (1553043_a_at) and CD33 (206120_at) extracted. The human GTEx data were analyzed in RStudio. Tissue type and transcription data in transcripts per million CD300LF and CD33 were extracted from all experiments in this data set.
Antibodies
Antibodies used for phenotyping can be found in the supplemental Methods.
Internalization assays
Details of internalization assays can be found in the supplemental Methods.
Generation of DCR-2-PBD and isotype-PBD
DCR-2 is a murine immunoglobulin G1 antibody that binds all extracellular forms of CD300f. DCR-2 and an isotype control antibody were conjugated to pyrrolobenzodiazepine (PBD) dimers by native cysteine chemistry via a cathepsin B–cleavable linker, MA-PEG4-VA-PBD (SET0212, Levena Biopharma). Briefly, monoclonal antibody (mAb) was reduced with 10 mM dithioerythritol to expose free thiols of interchain disulfides. The reduced antibody was reacted with a 10-fold molar excess of PBD linker (MA-PEG4-PBD) (10 mg/mL) for 3 hours before overnight dialysis into phosphate-buffered saline (PBS). The final drug-to-antibody molar ratios were 3.3 for DCR-2-PBD and 4.6 for isotype-PBD.
Cytotoxicity assays
Details of cytotoxicity assays can be found in the supplemental Methods.
CFU toxicity assay
Human CB or primary AML cells were plated at 2 × 104 cells/well in semisolid methylcellulose medium (MethoCult Classic, Stemcell Technologies) in a 24-well plate (Corning) were cultured at 37°C in 5% CO2 for 14 days when colony-forming units (CFUs) were scored. DCR-2-PBD or isotype-PBD was added to each well for continuous exposure. Three CB samples were tested with duplicate wells performed in each experiment. Primary AML cells were taken from PB samples with >90% blasts and performed in duplicate.
Activation marker expression and MoDC toxicity assays
Mouse xenograft assays
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (Australian BioResources) were housed under specific-pathogen-free conditions. In the subcutaneous model, 2 × 106 U937 cells were injected under the skin on the right flank. Tumors were measured daily with electronic calipers. When the mean volume of the tumors was >100 mm3, DCR-2-PBD, isotype-PBD, or PBS was injected intraperitoneally once. Mice were assigned so each cohort had a similar mean starting tumor volume (range, 130.3-137.2 mm3). Tumor volume was measured until >1000 mm3, when mice were euthanized.
For the BM HL-60 model, NSG mice were irradiated with 200 cGy 24 hours prior to IV administration of 5 × 106 HL-60 cells. On day 7, mice were injected with DCR-2-PBD, isotype-PBD, or PBS and monitored for disease progression and survival. Mice were euthanized when their clinical score was ≥4 or on day 70.
The humanized mouse model used CD34+ cells (>95% purity) isolated by positive selection (Miltenyi 130-046-702) according to the manufacturer’s instructions from 2 pooled CB samples. Each NSG mouse was injected with 100 000 CD34+ cells 4 hours after receiving a 150-cGy irradiation dose. Engraftment as determined by percentage of human CD45+ cells in venous blood was assessed at 12 weeks. Mice were assigned to treatment groups to have similar means of human CD45+ percentage in the PB (10.16% DCR-2-PBD cohort and 10.74% isotype-PBD cohort). Mice were then injected with 300 μg/kg of DCR-2-PBD or isotype-PBD. Mice were euthanized on day 7, and human CD45+ cells from BM (bilateral femurs and tibias) and 300 μl blood were enumerated and phenotyped by flow cytometry.
For primary AML xenografts, NSG mice were irradiated with 150 cGy 24 hours before 8 × 106 AML cells from CRGH11 (sample 10, supplemental Table 1) were injected IV. Once engraftment was established (>1% human CD45-positive events in PB), mice were given DCR-2-PBD or isotype-PBD. Mice were euthanized 6 days later, and the BM was harvested for enumeration and surface marker analysis by flow cytometry.
Flow cytometry
CD300f expression on primary AML and BM samples was performed on an Influx flow cytometer (BD Biosciences). AML cell line experiments were performed on an Accuri flow cytometer (BD Biosciences). Remaining assays were performed on a Fortessa LSR flow cytometer (BD Biosciences). Analysis, including TiSNE, was performed using FlowJo.
Statistical analysis
Statistical analysis was performed using Prism (GraphPad Software). Error bars correspond to standard error. Differences in means between 2 groups were assessed using Student t tests. Multiple-group tests were performed using analysis of variance with posttest comparisons. Differences in survival were assessed using a log-rank (Mantel-Cox) test. The combination index was calculated using Compusyn software.25
Results
CD300f is expressed on AML and HSPCs, but not outside of the hematopoietic system
To assess the distribution of CD300f, gene expression data were analyzed. As gentuzumab ozogamicin, which targets CD33, is the only approved AML therapeutic that binds a surface molecule, we compared expression of CD300f to CD33 in AML. We analyzed expression array data from patients with AML (n = 460) to compare CD33 and CD300LF (Figure 1A). The mean CD300LF expression level was significantly higher than CD33 (8.39 log2 vs 7.89 log2, P < .0001). There was a weak correlation in transcript expression between CD300LF and CD33 (r = 0.16, P = .004). There were no significant differences in CD300LF expression across the European LeukemiaNet risk groups (supplemental Figure 1A).
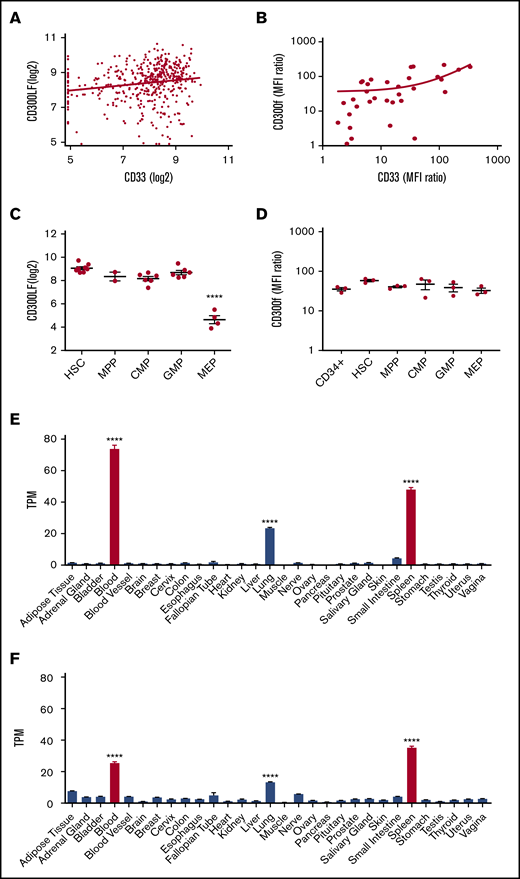
CD300f expression on AML cells, HSPCs, and organ expression. (A) CD33 is compared with CD300f by gene expression using linear regression analysis. (B) MFI ratios of CD33 and CD300f on patient-derived AML cells (n = 33) are compared with linear regression. (C) CD300f gene expression across HSPC subtypes (****P < .0001 megakaryocyte/erythroid progenitors vs all other subtypes). (D) MFI ratios of CD300f on HSPC from healthy BM. (E) CD300f gene expression by RNA-seq across multiple organ types from the human GTEx dataset (****P < .0001 blood, spleen or lung vs all organs). (F) CD33 gene expression by RNA-seq across multiple organ types (****P < .0001 blood, spleen, or lung vs all organs). Red bars indicate hematopoietic organs. TPM, transcripts per million.
CD300f expression on AML cells, HSPCs, and organ expression. (A) CD33 is compared with CD300f by gene expression using linear regression analysis. (B) MFI ratios of CD33 and CD300f on patient-derived AML cells (n = 33) are compared with linear regression. (C) CD300f gene expression across HSPC subtypes (****P < .0001 megakaryocyte/erythroid progenitors vs all other subtypes). (D) MFI ratios of CD300f on HSPC from healthy BM. (E) CD300f gene expression by RNA-seq across multiple organ types from the human GTEx dataset (****P < .0001 blood, spleen or lung vs all organs). (F) CD33 gene expression by RNA-seq across multiple organ types (****P < .0001 blood, spleen, or lung vs all organs). Red bars indicate hematopoietic organs. TPM, transcripts per million.
Protein expression of CD300f and CD33 was compared by flow cytometry (Figure 1B). There was no significant difference in mean fluorescence intensity (MFI) ratios, and there was a moderate to strong correlation in expression (r = 0.63, P = .0001). CD300f protein expression on the CD34+CD38− subset of primary AML showed no significant correlation with CD33 protein expression (supplemental Figure 1B). Single-cell expression was compared on a set of 9 AMLs using TiSNE, with a similar distribution seen for CD300f (supplemental Figure 1C) and CD33 (supplemental Figure 1D). Gene expression array data demonstrated CD300f expression across hematopoietic stem cells and myeloid hematopoietic progenitor cells (Figure 1C), with the lowest expression in megakaryocyte/erythroid progenitors (P < .0001). Protein expression by flow cytometry did not reveal any significant differences in MFI ratio across the groups of HSPCs. A standard gating strategy (supplemental Figure 3) was used to determine HSPC subsets. Protein expression on peripheral blood mononuclear cells showed CD300f expression on monocytes and neutrophils, but not lymphocytes (supplemental Figure 2). RNA sequencing (RNA-seq) data from the human GTx database of multiple organs were analyzed for potential off-target effects of anti-CD300f–based therapy. Both CD33 and CD300LF had significantly increased expression in the blood, spleen, and lungs compared with all other organs (P < .0001) (Figure 1E-F). Immunohistochemistry analysis by The Human Protein Atlas (https://www.proteinatlas.org/) indicates that CD33 and CD300f are expressed on lung macrophages, but not on pneumocytes.
Antibodies to CD300f are internalized upon binding
The mouse anti-human CD300f antibody, DCR-2 was assessed for its ability to be internalized by flow cytometry. It rapidly internalizes upon binding to HL-60 cells (Figure 2A). Internalization is significant at 30 minutes and is >50% at 180 minutes. Internalization at 30 minutes was confirmed by fluorescence microscopy on HL-60, CD14+ monocytes and primary AML cells (Figure 2B). The internalization data demonstrates that DCR-2 is a suitable mAb for ADC development.
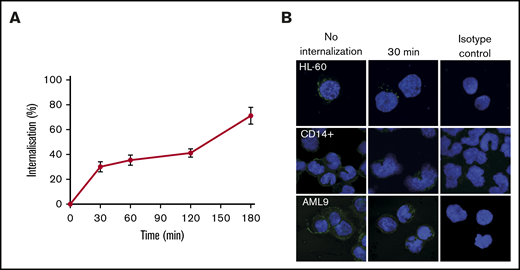
Internalization of DCR-2. (A) Internalization kinetics of DCR-2 on HL-60 (% internalized = relative MFI of total − relative MFI of surface staining). (B) Immunofluorescent microscopy of HL-60, CD14+ monocytes, and primary AML at 0 minutes (no internalization) and 30 minutes showing internalization of CD300f detected by LMIR-3 antibody. Isotype control shown is at 30 minutes. Images were taken at 63× magnification using a 63× HC PL APO CS2 NA 1.40 lens.
Internalization of DCR-2. (A) Internalization kinetics of DCR-2 on HL-60 (% internalized = relative MFI of total − relative MFI of surface staining). (B) Immunofluorescent microscopy of HL-60, CD14+ monocytes, and primary AML at 0 minutes (no internalization) and 30 minutes showing internalization of CD300f detected by LMIR-3 antibody. Isotype control shown is at 30 minutes. Images were taken at 63× magnification using a 63× HC PL APO CS2 NA 1.40 lens.
In vitro cytotoxicity of DCR-2-PBD
DCR-2 and an isotype control mAb were conjugated to a PBD toxin to assess potential cytotoxicity. The cytotoxicity of DCR-2-PBD and the isotype-PBD were tested against CD300f+ and CD300f− cell lines (Figure 3A). DCR-2-PBD killed the CD300f+ AML cell lines HL-60, U937, and THP-1 with 50% inhibitory concentrations (IC50s) in the low picomolar range (5.44, 6.74, and 29.39 pM, respectively). These IC50s are similar to those of other PBD-based ADCs.26,27 DCR-2-PBD has >200 pM IC50 against the CD300− lymphoma cell line Z-138. The isotype-PBD has an IC50 of >200 pM against HL-60 and U937.
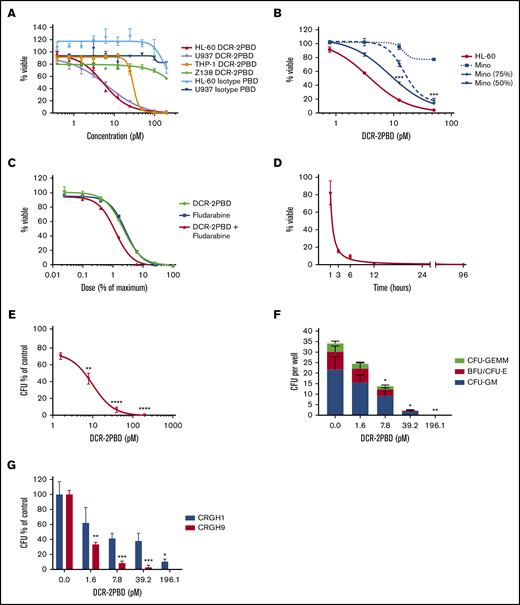
DCR-2-PBD in vitro cytotoxicity. (A) Inhibitory concentration curves on CD300f+ AML cell lines (HL-60, U937, and THP-1) and CD300f− lymphoma cell line (Z-138) using DCR-2-PBD or isotype-PBD (performed in triplicate). (B) Bystander killing assay using an CD300f− lymphoma cell line (Mino) and HL-60 either alone or in combination. In combination conditions, only the percentage of viable of Mino cells is shown (performed in triplicate) (***P < .001 Mino 50% at 25 and 12.5 pM, Mino 75% at 25 pM). (C) Combination DCR-2-PBD and fludarabine inhibitory concentration curves using HL-60 (performed in triplicate). (D) Time-dependent killing assay of DCR-2-PBD on HL-60, with final viability measured at 96 hours. (E) Total CFU inhibitor concentration curve with DCR-2-PBD (3 CB samples each performed in duplicate) (**P < .01 CFU inhibition at 7.8 pM, ****P < .0001 CFU inhibition at 39.2 and 196.1 pM). (F) Individual CFU subtype formation inhibition by DCR-2-PBD (*P < .05 CFU inhibition at 7.8 and 39.2 pM, **P < .01 CFU inhibition at 196.1 pM). (G) Individual AML CFU inhibition with DCR-2 PBD (performed in duplicate) (*P < .05 CFU inhibition at 196.2 pM for CRGH1, **P < .01 CFU inhibition at 1.6 pM for CRGH9, ***P < .001 CFU inhibition at 39.2 pM and 196.2 pM for CRGH9). BFU, burst-forming unit; GEMM, granulocyte, erythroid, macrophage, megakaryocyte; GM, granulocyte macrophage.
DCR-2-PBD in vitro cytotoxicity. (A) Inhibitory concentration curves on CD300f+ AML cell lines (HL-60, U937, and THP-1) and CD300f− lymphoma cell line (Z-138) using DCR-2-PBD or isotype-PBD (performed in triplicate). (B) Bystander killing assay using an CD300f− lymphoma cell line (Mino) and HL-60 either alone or in combination. In combination conditions, only the percentage of viable of Mino cells is shown (performed in triplicate) (***P < .001 Mino 50% at 25 and 12.5 pM, Mino 75% at 25 pM). (C) Combination DCR-2-PBD and fludarabine inhibitory concentration curves using HL-60 (performed in triplicate). (D) Time-dependent killing assay of DCR-2-PBD on HL-60, with final viability measured at 96 hours. (E) Total CFU inhibitor concentration curve with DCR-2-PBD (3 CB samples each performed in duplicate) (**P < .01 CFU inhibition at 7.8 pM, ****P < .0001 CFU inhibition at 39.2 and 196.1 pM). (F) Individual CFU subtype formation inhibition by DCR-2-PBD (*P < .05 CFU inhibition at 7.8 and 39.2 pM, **P < .01 CFU inhibition at 196.1 pM). (G) Individual AML CFU inhibition with DCR-2 PBD (performed in duplicate) (*P < .05 CFU inhibition at 196.2 pM for CRGH1, **P < .01 CFU inhibition at 1.6 pM for CRGH9, ***P < .001 CFU inhibition at 39.2 pM and 196.2 pM for CRGH9). BFU, burst-forming unit; GEMM, granulocyte, erythroid, macrophage, megakaryocyte; GM, granulocyte macrophage.
AML surface targets, including CD300f, can be heterogeneously expressed within AML cases, and we considered bystander killing advantageous for an effective ADC. We investigated bystander killing by comparing the viability of the CD300f− lymphoma cell line Mino cultured on its own with Mino cultured mixed with HL-60 in the presence of DCR-2-PBD (Figure 3B). There was a significant reduction in viability in the 75% Mino condition with DCR-2-PBD at 25 pM (P = .0002) compared with the Mino-only control. In the 50% Mino condition, there was a significant reduction in viability in both the 25 pM (P < .0001) and 12.5 pM (P = .0004) DCR-2-PBD concentrations. The reduction in Mino cells when combined with HL-60 confirms the ability of DCR-2-PBD to generate bystander killing.
Fludarabine is used in many allo-HSCT regimens to facilitate donor cell engraftment by depleting host lymphocytes. Fludarabine combined with PBD demonstrates synergistic cytotoxic properties.28 An anti-CD300f therapeutic would deplete recipient HSPCs, but not host lymphocytes, as part of a conditioning regimen. Given these complementary roles, we assessed a synergistic relationship between DCR-2-PBD and fludarabine. Cytotoxicity of HL-60 (Figure 3C) and THP-1 (data not shown) was greater in the presence of both DCR-2-PBD and fludarabine than either alone, confirming a synergistic relationship with a combination index of 0.82 and 0.76, respectively. We assessed the exposure time required for DCR-2-PBD to cause cytotoxicity on the HL-60 cell line (Figure 3D). At 12 hours, >98% of HL-60 proliferation was inhibited, while at 24 and 96 hours, inhibition was >99%.
We used a CFU assay to test DCR-2-PBD activity against HSPC and primary AML. Significant reductions in total CFUs were seen at 7.84 (P = .0033), 39.21 (P < .0001), and 196.1 pM (P < .0001) (Figure 3E). All major CFU subtypes were inhibited at the 32.9 and 196.1 pM concentrations by DCR-2-PBD (Figure 3F). Two primary AML samples were tested: CRGH1 with low CD300f expression (MFI ratio of 2.6) and CRGH9 with a high CD300f expression (MFI 109.9) (Figure 3G). Significant toxicity was demonstrated against CRGH1 at 196.1 pM (P = .036). Significant toxicity was seen in CRGH9 at all dose levels (P < .001).
CD300f is expressed on myeloid APCs; therefore, depletion of CD300f-expressing cells may impair recipient antigen presentation.29 We examined if DCR-2-PBD would reduce myeloid APC cell numbers, prevent activation, or have a functional inhibitory effect (supplemental Figure 4). MoDCs were used to test the ability of DCR-2-PBD to deplete APCs. DCR-2-PBD did not significantly reduce MoDC numbers. PBD has demonstrated cytotoxicity against quiescent and dividing cells but to our knowledge has not been previously trialed on terminally differentiated myeloid cells.30 The impact of DCR-2-PBD on myeloid APC activation was assessed by incubating APCs with DCR-2-PBD followed by lipopolysaccharide. DCR-2-PBD did not inhibit the expression of activation markers on Lineage− HLADR+ CD11c+ myeloid dendritic cells. DCR-2-PBD did not prevent T-cell activation in a 1-way mixed lymphocyte reaction.
DCR-2-PBD prolongs the survival of mouse AML cell line models
DCR-2-PBD in vivo function was tested in subcutaneous and BM cell line models. U937 was injected subcutaneously in NSG mice. Mice were injected with a single dose of PBS, isotype-PBD (150 or 300 μg/kg) or DCR-2-PBD (150 or 300 μg/kg) (Figure 4A), and mice were monitored for tumor growth. Tumor growth was delayed, and survival was significantly increased in both the 150 μg/kg group (P = .0019) and 300 μg/kg (P = .0017) of DCR-2-PBD compared with the isotype-PBD (300 μg/kg) group. A second model used irradiated mice that were IV injected with HL-60, which led to heavy BM infiltration, met clinical end points at a median of day 23. A single injection of DCR-2-PBD (300 μg/kg) significantly increased survival (P = .0058) compared with isotype-PBD (Figure 4B). Six of 8 animals treated with DCR-2-PBD survived to the experiment end point. No human cells could be detected in mice surviving until day 70, and the range of human CD45+ cells in all other mice was 76% to 88% at the time of euthanasia.
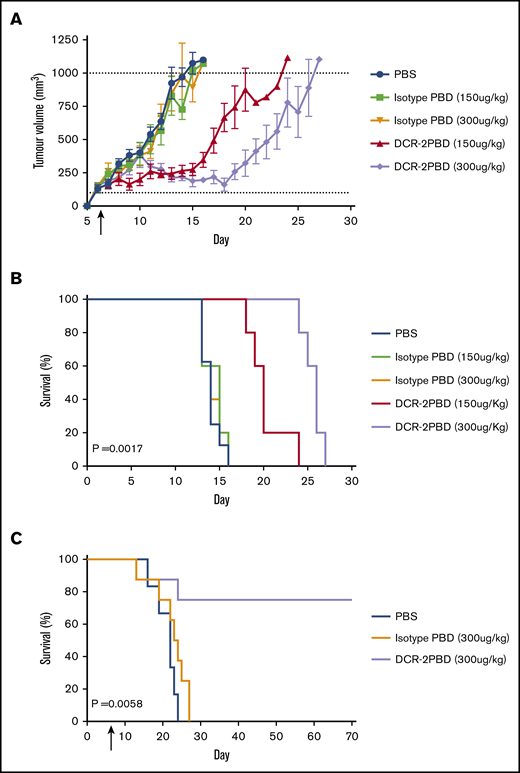
DCR-2-PBD prolongs survival in AML cell line mouse models. (A) U937 subcutaneous tumor volume of mice treated with a single injection on day 6 with either PBS (n = 8) or isotype-PBD (n = 5 both groups) or DCR-2-PBD (n = 5 both groups). (B) Survival data from mice injected with U937 subcutaneously (P = .0017 DCR-2-PBD 300 μg/kg compared with isotype-PBD 300 μg/kg). (C) Mice injected with HL-60 IV then treated day 7 with PBS (n = 6), isotype-PBD (n = 8), or DCR-2-PBD (n = 8) (P = .0058 DCR-2-PBD vs isotype-PBD).
DCR-2-PBD prolongs survival in AML cell line mouse models. (A) U937 subcutaneous tumor volume of mice treated with a single injection on day 6 with either PBS (n = 8) or isotype-PBD (n = 5 both groups) or DCR-2-PBD (n = 5 both groups). (B) Survival data from mice injected with U937 subcutaneously (P = .0017 DCR-2-PBD 300 μg/kg compared with isotype-PBD 300 μg/kg). (C) Mice injected with HL-60 IV then treated day 7 with PBS (n = 6), isotype-PBD (n = 8), or DCR-2-PBD (n = 8) (P = .0058 DCR-2-PBD vs isotype-PBD).
DCR-2-PBD depleted both primary HSPCs and AML cells in mouse xenografts
Further proof of principle for an anti-CD300f ADC to contribute to allo-HSCT conditioning was demonstrated using DCR-2-PBD to selectively deplete HSPCs and myeloid cells in a humanized mouse model. Humanized mice injected with a single dose of DCR-2-PBD (300 μg/kg) had a significant depletion in the mean of both total CD34+ (0.29 × 105 vs 9.42 × 105, P = .001) and total primitive CD34+CD38−CD90+ cells (0.05 × 104 vs 5.54 × 104, P = .008) compared with isotype-PBD–treated groups (Figure 5A). The difference was also significant as a percentage of human CD45+ cells for both the CD34+ (3.09% vs 20.62%, P < .0001) and CD34+CD38−CD90+ populations (0.16% vs 2.17%, P = .008). The selectivity of DCR-2-PBD compared with isotype-PBD was demonstrated by depleting the myeloid cells (0.31 cells/μL vs 14.89 cells/μL, P = .0002), but not lymphocytes (208.5 cells/μL vs 263 cells/μL), which are longer lived and do not express CD300f, from PB samples (Figure 5B).
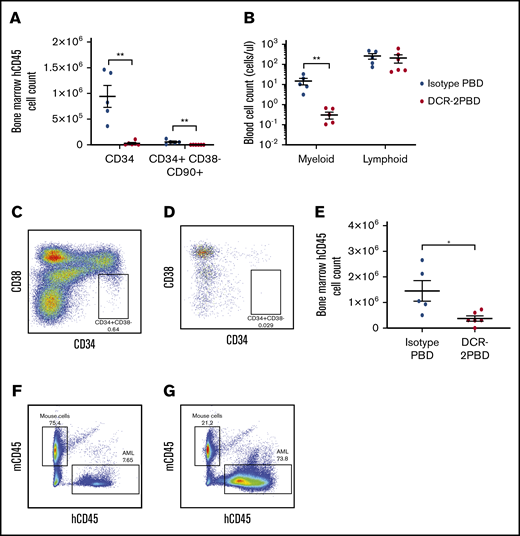
DCR-2-PBD depletes HSPCs in vivo. (A) Total cell count of CD34+ and CD34+ CD38−CD90+ cells in humanized NSG mouse BM 7 days after injection of DCR-2-PBD (n = 6) or isotype-PBD (n = 5) (**P < .01 reduction in CD34+ and CD34+ CD38−CD90+ cells in the DCR-2-PBD cohort compared with the isotype control cohort). (B) Cell count per microliter of blood from DCR-2-PBD and isotype-PBD treated humanized mice (**P < .01 reduction in myeloid cells in PB of DCR-2-PBD cohort compared with isotype-PBD cohort). (C-D) Representative plots of human CD45+ cells in the BM of a control humanized mouse (C) and a DCR-2-PBD treated humanized mouse (D). (E) BM enumeration of primary AML 6 days after injection of mice treated with isotype-PBD and DCR-2-PBD (n = 6 DCR-2-PBD, n = 5 isotype-PBD) (*P < .05 reduction of primary AML cells in BM, DCR-2-PBD cohort compared with the isotype-PBD cohort). (F-G) Representative plots of mouse CD45 vs human CD45 cells in mice engrafted with primary AML treated with isotype-PBD (F) and DCR-2-PBD (G).
DCR-2-PBD depletes HSPCs in vivo. (A) Total cell count of CD34+ and CD34+ CD38−CD90+ cells in humanized NSG mouse BM 7 days after injection of DCR-2-PBD (n = 6) or isotype-PBD (n = 5) (**P < .01 reduction in CD34+ and CD34+ CD38−CD90+ cells in the DCR-2-PBD cohort compared with the isotype control cohort). (B) Cell count per microliter of blood from DCR-2-PBD and isotype-PBD treated humanized mice (**P < .01 reduction in myeloid cells in PB of DCR-2-PBD cohort compared with isotype-PBD cohort). (C-D) Representative plots of human CD45+ cells in the BM of a control humanized mouse (C) and a DCR-2-PBD treated humanized mouse (D). (E) BM enumeration of primary AML 6 days after injection of mice treated with isotype-PBD and DCR-2-PBD (n = 6 DCR-2-PBD, n = 5 isotype-PBD) (*P < .05 reduction of primary AML cells in BM, DCR-2-PBD cohort compared with the isotype-PBD cohort). (F-G) Representative plots of mouse CD45 vs human CD45 cells in mice engrafted with primary AML treated with isotype-PBD (F) and DCR-2-PBD (G).
We demonstrated the efficacy of DCR-2-PBD against primary AML by injecting mice with a single dose of 300 μg/kg DCR-2-PBD or isotype-PBD and enumerating BM engraftment on day 7. DCR-2-PBD significantly depleted the human AML cells residing in the BM compared with isotype control when measured by mean total enumeration (0.37 × 106 vs 1.45 × 106P = .019) (Figure 5C-E). DCR-2-PBD significantly reduced the AML cells as a mean percentage of total cells (Figure 5F) compared with isotype-PBD (12.32% vs 38.92%, P = .03).
Discussion
The ideal way to use the next generation of surface-molecule–targeted therapy remains unclear. Avoiding an allo-HSCT would be ideal, but unless a true AML-specific antigen without expression on HSPCs is validated, hematological toxicity will remain unacceptably high. CD300f has been explored as an AML target, but a clear method on how to best use an anti-CD300f therapeutic has been lacking.17,18 Our work has validated CD300f as an ideal target for a novel conditioning agent in AML.
CD300f has an expression level and distribution comparable to CD33 both between and within AML cases. It is also unlikely to have significant expression outside the hematopoietic system. CD300f efficiently internalizes upon antibody binding, making it an excellent candidate for ADC development. DCR-2-PBD demonstrated specific in vitro toxicity of AML cell lines and HSPCs. The PBD component allows for rapid cytotoxicity and bystander killing. DCR-2-PBD prolonged survival in AML cell line models as well as depleted primary AML cells and HSPCs in vivo.
Alternatives to traditional chemotherapy-based treatments for AML include ADCs and chimeric antigen receptor T cells. As AML is derived from HSPCs, these cells share many surface membrane molecules, making the choice of a target difficult.31 Although gentuzumab ozogamicin had setbacks prior to its current listing, it provides evidence that an ADC may alter the course of AML in some patients.32 Vadastuximab talirine is an ADC that targets CD33 coupled to a PBD, which induced complete remission and MRD negativity in older patients with AML, but the maximum tolerated dose was limited by hematologic toxicity with BM aplasia.33 Nonhematologic toxicity was minimal. The marked hematologic toxicity demonstrated suggests that vadastuximab talirine targeted both AML and HSPCs. Vadastuximab talirine prior to allo-HSCT demonstrated tolerability and good outcomes for the small number of patients involved, suggesting no permanent effect of the PBD toxin limiting engraftment.34 The development of vadastuximab talirine ceased after an increased number of deaths due to infections were observed in the phase 3 CASCADE study.35 Incorporating an ADC into a transplant strategy may limit hematologic toxicity
We have shown that PBD, when internalized on an anti-CD300f antibody, causes significant cytotoxicity even with a brief exposure, which is ideal for a conditioning agent. A limitation with ADC-based conditioning is that the half-lives of most antibodies would endanger donor cells. Alternative strategies to shorten the half-life of an antibody by removing the FcRn recognition site or using an antibody fragment (ie, antigen-binding fragment) would address these concerns.36,37 A DCR-2–based therapeutic modified in this way will make such a conditioning approach possible.
CD300f has the potential to be variably expressed within a single case of AML, as this is common for many target antigens, including CD33.38 We have sought to mitigate this by employing a toxin with a bystander effect. The bystander effect of ADCs can be seen with brentuximab vedotin, which can induce a clinical response even in tumors with minimal expression.39 The ability of a toxin to kill bystander cells would enhance responses in heterogeneous tumors but also increase the potential for off-target effects. Given the inherent heterogeneity and plasticity of AML surface expression, choosing a toxin with a bystander effect, like we have demonstrated with DCR-2-PBD, will significantly increase efficacy. An immunosuppressive agent must combined with CD34+ depletion to facilitate engraftment. Fludarabine was found to be a natural partner to DCR-2-PBD given its widespread use in conditioning and synergistic properties with PBD.28
CD300f has some significant advantages to other molecules being explored in targeted conditioning. ADCs targeting CD45 and CD117 have been investigated for AML conditioning.40 While CD45 may contribute to both lymphodepletion and the reduction in healthy CD34+ cells and residual AML, it is expressed at low levels in AML and is not efficiently internalized.41 A higher total dose of ADC may overcome inefficient internalization, but this would increase the prospect of free toxin causing nonspecific toxicity. Anti-CD117 ADCs significantly reduce HSPCs in animal models.42 A major concern targeting CD117 with a potent ADC is its significant expression outside of the hematopoietic system, which raises the possibility of increased toxicity.43 The even distribution across the major HSPC subtypes would likely make CD300f a more efficient conditioning agent compared with current AML targets being studied, which often have variable HSPC expression.
We have demonstrated that CD300f is expressed on AML cells and HSPCs, and it is unlikely to be expressed significantly on cells of nonhematopoietic origin. CD300f is expressed on monocyte derived cells such as pulmonary macrophages and microglia in non-hematopoietic tissue.44-46 While this may suggest the potential for “off-target” toxicities, these same cells express CD33, and no severe nonhematologic toxicities were noted with the development of vadastuximab talirine.33 Despite this, a major limitation of this work is the examination of off-target ADC effects. The mouse anti-human CD300f ADC does not bind mouse CD300f, and therefore, mouse xenogeneic models would not be appropriate to examine nonhematopoietic toxicity. Any depletion of HSPCs may induce neutropenia given the very short half-life of neutrophils, regardless of the expression of the target on neutrophils themselves. We would expect an anti-CD300f therapeutic to induce neutropenia, but other ADCs used in transplant models have avoided neutropenia altogether while maintaining immunity, even when the target is expressed on neutrophils.14
We also investigated the impact of an anti-CD300f ADCs on host APCs. We predict the lack of reduction or effectiveness of myeloid APCs is due to inherent resistance to PBD as they are terminally differentiated. Tissue inflammation caused by nonspecific conditioning agents has a role in the development of graft-versus-host disease, and an ADC conditioning backbone may significantly alter the posttransplant immune landscape.47 A DCR-2-PBD–based therapeutic would still require a lymphodepleting agent, which may mitigate any potential benefits of limiting tissue damage by removing alkylating agents or radiation. There are now multiple groups developing targeted conditioning, and the impact on graft-versus-host disease and graft-versus-leukemia needs to be studied further.
With the current range of AML targets, an allo-HSCT will be required as part of novel AML treatments to allow the use of highly potent therapies targeting myeloid antigens such as ADCs and chimeric antigen receptor T cells. An advantage of adapting these therapies to become part of transplant conditioning is that it may allow for advancements in transplantation outside of AML. The use of novel less toxic, specific conditioning agents will broaden their application from AML into transplantation for MDS, with the possibility of reducing toxicity in a cohort of patients that traditionally suffer significant morbidity and mortality from allo-HSCT. Recent advances in antibody-based therapeutics have presented the possibility of targeted conditioning agents.13,48 Clinical trials have begun with unconjugated antibodies as conditioning agents (NCT02963064). The potent effect demonstrated by DCR-2-PBD against healthy HSPCs suggests that therapeutic derivatives targeting CD300f may be incorporated into conditioning for nonmalignant disorders of erythropoiesis or immunodeficiency either as part of an allo-HSCT or a gene-modified autologous HSCT.
This work validates CD300f as a specific target that merits further exploration in targeted conditioning therapy. Testing across a greater number of primary AML samples and implementation of transplant models will be required.
For data sharing, please e-mail the corresponding author.
Acknowledgments
The authors acknowledge Elizabeth Newman and Christine Fong (Department of Haematology Concord Repatriation General Hospital) for the collection of venous and BM samples, the Sydney Cord Blood Bank for provision of CB, and Donna Bonnici for administrative assistance.
Authorship
Contribution: E.A. and G.J.C. designed the study and planned the experiments; E.A., P.A.S., R.E.G., M.R., A.R., A.H.M., T.-H.L., K.K., S.S., and G.A.P. performed experiments; E.A., P.A.S., R.E.G., and G.J.C. analyzed data; E.A. and G.J.C. wrote the manuscript; and P.A.S., R.E.G., S.S., C.E.B., P.J.H., and S.R.L. assisted with revision of the manuscript.
Conflict-of-interest disclosure: G.J.C. is a director of DendroCyte BioTech Pty Ltd. G.J.C. and R.E.G. are listed as inventors on patents protecting DCR-2. The remaining authors declare no competing financial interests.
Correspondence: Georgina J. Clark, Dendritic Cell Research, ANZAC Research Institute, Gate 3 Hospital Rd, Concord, NSW 2139, Australia; e-mail: georgina.clark@sydney.edu.au.

