

Key Points
Targeting fibrin formation directly is a novel approach to reduce microvascular thrombosis in systemic inflammation.

Abstract
Systemic inflammation can lead to coagulopathy and disseminated intravascular coagulation (DIC). In prior studies, the recombinant A2 domain of human von Willebrand factor (VWF; A2 protein) attenuated DIC and decreased mortality in lipopolysaccharide (LPS)-treated mice. Here, we performed studies to dissect the mechanism by which the A2 protein moderates DIC. We used confocal microscopy to analyze the fibrin clot structure in plasma from healthy humans and endotoxemic mice, turbidity assays to examine fibrin polymerization, and a murine model for LPS-induced DIC and introduced a loss-of-function mutation into the A2 protein for fibrin. The mutation of the residue E1567 located in the α2 helix of the folded A2 domain of VWF inhibited binding activity for fibrin, possibly mapping a novel region containing a putative binding site for fibrin. The A2 protein increased the initial rate of change of fibrin polymerization, intercalated into the fibrin network, and modified the resultant clot structure in vitro. Furthermore, ex vivo experiments using plasma from mice with endotoxemia treated with the A2 protein revealed an increased rate of fibrin formation and an altered clot structure as compared with plasma from nontreated sick animals. Moreover, and in contrast to the A2 mutant, the A2 protein improved survival and reduced fibrin deposition and microvascular thrombosis in mice with endotoxemia-induced DIC. Importantly, in vivo and in vitro studies indicated that the A2 protein did not affect experimental thrombosis. Thus, we provide evidence for a novel treatment to attenuate systemic inflammation-induced coagulopathy/DIC via targeting fibrin formation, without an increased risk for bleeding.
Introduction
Persistent systemic inflammation can activate the coagulation and thrombotic pathways, leading to a prothrombotic and antifibrinolytic state, as seen in endotoxemia, sepsis, and bacteremia. The resultant widespread fibrin deposition in small to midsize blood vessels leads to organ ischemia and dysfunction.1 The presence of widespread fibrin deposition and thrombosis in the microvasculature is a hallmark of disseminated intravascular coagulation (DIC), which can occur in 29% to 50% of septic patients and is associated with increased mortality.2-4 Although the treatment of sepsis includes source control, antibiotics, and hemodynamic resuscitation, no therapy currently exists for sepsis-induced DIC other than supportive care.
Fibrinogen plays an essential role in hemostasis and thrombosis. During coagulation, thrombin converts fibrinogen into fibrin, forming the insoluble end product of the coagulation pathway (as reviewed by Kattula et al5 ). A number of conditions, including coagulation factors, plasma components, blood cells, and blood flow, contribute to the formation, structure, and stability of the resultant fibrin clot (as reviewed by Chandrashekar et al6 ). It is well appreciated that alterations in the fibrin clot profile are directly associated with different clinical pathologies, including conditions associated with bleeding and thrombosis.7-12 Therefore, the development of interventions to modify fibrin clot structure and stability to prevent pathologic hemorrhage and thrombosis in systemic inflammation is an unmet medical need.
We have previously shown that the recombinant A2 domain of human von Willebrand factor (VWF), the A2 protein, effectively binds to fibrin and reduces platelet clot formation in flowing blood. Importantly, the A2 protein decreased mortality from 60% to 0% and attenuated disseminated microvascular thrombosis in an endotoxemia-induced DIC murine model.13 However, it was unclear whether A2 protein–fibrin binding was the main mechanism by which the A2 protein led to improved survival in our lipopolysaccharide (LPS)-induced DIC murine model. Therefore, the goal of our current study was to elucidate further the molecular mechanism by which the A2 protein improves survival and attenuates DIC in endotoxemic mice. Our results show that the A2 protein increases the initial rate of change of fibrin polymerization, intercalates into the fibrin network, and modifies the resultant clot structure in vitro. Furthermore, ex vivo experiments employing plasma from mice with endotoxemia treated with the A2 protein revealed an increased rate of fibrin formation as compared with plasma from nontreated sick animals. Lastly, mutation of the residue E1567 located in the α2 helix of the folded A2 domain of VWF inhibited binding activity for fibrin, suggesting that this residue might be part of the fibrin interaction site.
Materials and methods
Animal studies
All studies were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the institutional animal care and use committees of Baylor College of Medicine.
Reagents
The loss-of-function A2 mutant was constructed by introducing the E1567A mutation into the vector pQE-9/VWF-A2 using the QUIKCHANGE II XL site-directed mutagenesis kit (Stratagene, CA). We introduced the mutation by polymerase chain reaction using the following primers harboring the mutation: 5′ primer-GCAGCGGGTGCGAGCGATCCGCTACCAGG and 3′ primer-CCTGGTAGCGGATCGCTCGCACCCGCTGC. The recombinant A2 E1567A mutant was purified like wild-type (WT) A2 protein. Purified recombinant proteins (WT A2 and A2 E1567A mutant) were confirmed and validated as described.14 Structural integrity of the purified proteins was assessed by using the monoclonal anti-A2 antibody and circular dichroism thermal unfolding as described.15 Human fibrinogen was obtained from Calbiochem, and the recombinant human vimentin and monoclonal anti-human VWF-A2 domain (anti-A2) antibody were purchased from R&D Systems. The LPS (0111:B4) and antihistidine–horseradish peroxidase (HRP) conjugate antibody were obtained from Sigma. The recombinant A1 domain protein was produced as described previously.13,16
Imaging the fibrin clot structure
To obtain human blood, informed consent was provided based on the recommendations of the Declaration of Helsinki. Approval was attained from the Baylor College of Medicine Institutional Review Board for these studies. The effect of the A2 protein on fibrin clot structure was evaluated using platelet-poor plasma collected from human participants. Three-dimensional reconstructions of the fibrin clot structure were obtained by supplementing the plasma with 2% (WT/WT) human fibrinogen conjugated to Alexa Fluor 647 (Thermo Scientific) and initiating clot formation with the addition of 1 U of thrombin (EMD Millipore) in the presence of 2.4 mM of calcium. In studies with endotoxemic murine plasma, the A2 protein was injected intraperitoneally (IP) in vivo. Murine platelet-poor plasma was supplemented with 4% (WT/WT) human fibrinogen conjugated to Alexa Fluor 647 and clot formation initiated with 4 U of thrombin. In both instances, the plasma was transferred to a 35-mm glass-bottom dish (MatTek) and incubated at 37°C for 2 hours to allow clot formation, which was then imaged by laser scanning confocal microscopy. In some experiments, the A2 protein was fluorescently labeled through conjugation of the protein to Alexa Fluor 488 (Thermo Scientific). In these studies, the proteins were added and mixed with the whole blood before initiation of clot formation with the addition of thrombin. The whole blood was transferred to a 35-mm glass-bottom dish (MatTek) and incubated at 37°C for 2 hours to allow the clot to form. The whole blood was first fixed overnight and cleared using the c-clot protocol as previously described,17 before being imaged by laser scanning confocal microscopy.
Fibrin polymerization assays
Fibrin formation was evaluated by turbidity assay using healthy human plasma or endotoxemic plasma as described.13 The progression of fibrin clot formation was evaluated by tracking turbidity using a spectrophotometer set to λ 405 nm. Fibrinolysis was performed by mixing human healthy plasma with the A2 protein (2.0 or 4.0 μM) or saline in the presence of 150 ng/mL of tissue plasminogen activator (Cathflo Activase) and 0.1 U of human thrombin. The progression of fibrin clot formation and fibrinolysis was evaluated by tracking turbidity using a spectrophotometer set to λ 405 nm.
Binding assays
The binding of the WT A2 and A2 E1567A mutant to fibrin, vimentin, and the A1 domain protein were performed using enzyme-linked immunosorbent assay as we previously described.13,18,19 To convert fibrinogen to fibrin, 0.1 U/mL of thrombin (Sigma) was incubated in wells coated with fibrinogen (5.0 µg/mL) for 45 minutes at 37°C. After incubation and washing, increasing concentrations of the A2 variants were added into the wells. The bound proteins were detected using monoclonal antihistidine-HRP conjugate. Anti-A2 antibody was used to detect A2 variants bound to either vimentin or the A1 domain protein followed by a secondary HRP conjugate antibody.
SPOT synthesis of the A2 peptide sequence
SPOT synthesis was used to identify key residues within the A2 protein that are important for fibrin binding.20 Although the entire A2 sequence was evaluated, overlapping peptide sequences of 18 residues in length were synthesized on cellulose membranes by automated SPOT synthesis using a MultiPep RS (Intavis, Bergisch Gladback, Germany) as described elsewhere.21 After synthesis, the membranes were soaked for 10 minutes in methanol, followed by 2 10-minute washes in phosphate-buffered saline (PBS; pH, 7.4) before incubation in blocking buffer overnight at 4°C with gentle rocking. The following day, the membrane was washed 3 times for 10 minutes each with PBS, 0.05% Tween 20 (PBS-T), and 1% bovine serum albumin (BSA). Fibrin (Sigma) was biotinylated and dialyzed against PBS. The biotinylated fibrin was diluted to a concentration of 75 µg/mL in PBS-T and 1% BSA and incubated with the membrane for 2 hours with gentle shaking at room temperature. After incubation, the membrane was washed 3 times with PBS-T and 1% BSA for 10 minutes each and incubated with avidin-HRP diluted in PBS-T and 1% BSA for 1 hour at room temperature with gentle shaking. The membrane was washed 3 times with PBS-T and 1% BSA and developed with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) for 1 minute and then exposed to autoradiography film. The key residues were identified as described,22 and the 3-dimensional structure representation was created using UCSF Chimera software.
Flow assays
Dishes were coated with collagen type III and perfused with whole blood from healthy donors containing vehicle control buffer or the WT A2 protein (4.0 μM). Perfusion assays were carried out as we have described elsewhere.18 Platelets were observed with phase contrast objectives, recorded by videomicroscopy, and analyzed by using the MacBiophotonics ImageJ program.
Intravital microscopy
Preparation of the cremaster muscle of male C57Bl/6J mice and photoactivation to induce thrombosis and analyses were performed as described by our group.23,24 Briefly, after equilibration, fluorescein isothiocyanate–dextran (150 kD; 10 mL/kg of a 5% solution) was injected via the venous catheter and allowed to circulate for ∼10 minutes. Thereafter, venular diameter was measured (Image 1.6; National Institutes of Health public domain software) as well as mean blood cell velocity (Vdoppler, using an optical Doppler velocimeter; Cardiovascular Research Institute, Texas A&M University). Venular wall shear rate (γ) was calculated as 8(Vdoppler/1.34)/diameter. After those measurements, light/dye-induced injury was begun by exposing ∼100 µm of vessel length to epiillumination, with a 175-W xenon lamp (Λ LS; Sutter, Novato, CA) and a fluorescein filter cube (HQ-FITC; Chroma, Bellows Falls, VT). Excitation light was monitored daily (IL 1700 Radiometer, SED-033 Detector; International Light, Peabody, MA) and maintained at 0.6 W/cm2. Epiillumination was applied continuously, and the following times were recorded: time of onset of platelet aggregates and time of flow cessation, for at least 60 seconds. Five minutes before thrombus induction, animals received a single IV bolus of the A2 protein at a dosage of 4 mg/kg (n = 8). Control animals received equivalent volumes of physiological saline (n = 8).
LPS-induced DIC murine model and histology
Briefly,13 mice (C57BL/6; age 10-12 weeks old) were injected IP with LPS (30 mg/kg). The WT A2 (n = 10) or A2 E1567A (n = 12) mutant or saline (n = 6) was injected IP at a concentration of 4 mg/kg 1.5 hours after the LPS injection. Saline was used as positive control. Mice that did not receive either injection were used as negative controls. At 24 hours after LPS injection, kidneys were harvested from mice. These organs were processed using the services of the Comparative Pathology Laboratory of Baylor College of Medicine. Microvascular fibrin-rich thrombi in paraffin-embedded kidney tissues were studied by immunostaining using the polyclonal fibrinogen antibody (Dako, Carpinteria, CA). Histology images were analyzed using an Olympus FV3000 microscope.
Statistics
We used Prism by GraphPad Prism 8 software (San Diego, CA) and SigmaPlot by Systat Software, Inc. (San Jose, CA), to perform our statistical analyses, including 2-factor repeated-measures analysis of variance, nonlinear fit regression, and the Student t test. P < .05 was considered statistically significant.
Results
Identification of a putative binding site for fibrin in the A2 domain of VWF
We used the peptide SPOT array technique20 to identify the putative contact sites for fibrin within the A2 domain structure. To this end, 18meric overlapping peptides derived from the amino acid sequence of the A2 domain (G1481-R1668) were directly synthesized on a cellulose membrane and probed for binding to fibrin (supplemental Figure 1). After analyzing the intensity of the binding spots, the resultant residues were mapped onto the crystal structure of the A2 domain (Figure 1A).25 The regions colored in green (α2-helix) and magenta (α6-helix) most likely form the putative contact sites for fibrin, although the segment colored in blue (β1-strand) could contribute if the A2 protein is unfolded before the binding to fibrin. Among the A2 mutants constructed (not shown), the purified A2 (E1567A) mutant (supplemental Figure 2A) exhibited a much lower binding affinity for fibrin than that of the WT A2 protein (half maximal binding constant, 1.03 ± 0.079 µM vs 0.06 ± 0.004 µM, respectively; Figure 1B). Note that the peptide sequence containing the amino acid residue E1567 on the cellulose membrane was readily detected by fibrin (Figure 1; supplemental Figure 1). Another feature of this A2 mutant is that in comparison with the WT A2 protein, the E1567A mutation did not alter the overall structure of the recombinant A2 protein, as demonstrated by studies using a monoclonal antibody (Figure 1C) and circular dichroism thermal unfolding (Figure 1D). Furthermore, the A2 mutant bound to both the recombinant A1 domain of VWF and vimentin (supplemental Figure 2B-C, respectively), comparably to the WT A2 protein. Thus, the E1567A mutation did not alter the overall A2 protein structure and specifically impaired the interaction with fibrin without affecting the binding affinity of either the A1 domain of VWF or vimentin. The residue E1567 apparently forms part of the contact region for fibrin in the A2 protein.

Identification of fibrin contact regions in the A2 domain of VWF. (A) Crystal structure of the A2 domain. Magenta, green, and blue colors show regions containing putative binding sites for fibrin. The amino acid residue E1567 (green) analyzed in this study is depicted as a point of reference; red shows the location of the ADAMTS-13 cleavage site. (B) Increasing concentrations of either the WT A2 protein or A2 mutant were incubated with immobilized thrombin-generated fibrin in microtiter wells. The A2 mutant had significantly lower binding activity for fibrin (half-maximal binding, 1.03 ± 0.079 µM) than the WT A2 protein (half-maximal binding, 0.06 ± 0.004 µM). Each point in the graph represents the mean ± standard error of the mean of 3 determinations. Use of monoclonal antibody (C) and circular dichroism thermal unfolding (D) shows that the overall structure of the A2 protein was not altered by the mutation E1567A. (D) Assessment of 2 different batches of the WT A2 protein. *P < .05. Abs, absorbance; n.s., not significant.
Identification of fibrin contact regions in the A2 domain of VWF. (A) Crystal structure of the A2 domain. Magenta, green, and blue colors show regions containing putative binding sites for fibrin. The amino acid residue E1567 (green) analyzed in this study is depicted as a point of reference; red shows the location of the ADAMTS-13 cleavage site. (B) Increasing concentrations of either the WT A2 protein or A2 mutant were incubated with immobilized thrombin-generated fibrin in microtiter wells. The A2 mutant had significantly lower binding activity for fibrin (half-maximal binding, 1.03 ± 0.079 µM) than the WT A2 protein (half-maximal binding, 0.06 ± 0.004 µM). Each point in the graph represents the mean ± standard error of the mean of 3 determinations. Use of monoclonal antibody (C) and circular dichroism thermal unfolding (D) shows that the overall structure of the A2 protein was not altered by the mutation E1567A. (D) Assessment of 2 different batches of the WT A2 protein. *P < .05. Abs, absorbance; n.s., not significant.
A2 protein influences the initial change of the fibrin formation rate in human plasma
Next, we employed turbidity assays to examine the hypothesis that the A2 protein influences the kinetics of fibrin polymerization in plasma from healthy human donors. The assays were conducted using plasma containing increasing concentrations of the A2 protein. Figure 2A-B shows that the A2 protein significantly reduced the time (ie, increased the rate) to reach the maximum (optical density absorbance) turbidity in a dose-dependent manner. Similarly, the initial rate of change increased as the A2 concentrations increased (supplemental Table 1). The initial rate of change was determined from the slope of the line at the midpoint between initial baseline and maximum absorbance as described.26 In contrast, the A2 (E1567A) mutant did not affect the rate of fibrin polymerization (supplemental Figure 3A-B), suggesting that the A2 protein influences the rate of fibrin formation.
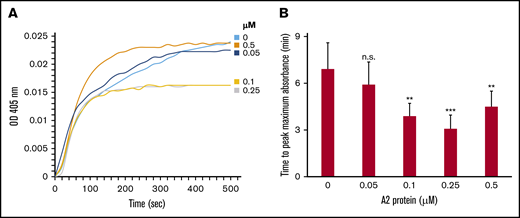
The A2 protein modulates the rate of fibrin formation in vitro. (A) Fibrin was formed from using 10% healthy human plasma and vehicle control or the A2 protein (0-0.5 μM), and turbidity was measured at λ 405 nm. The tracings shown are the average of 4 separate experiments. The baseline for fibrin polymerization in different experimental groups is normalized to time 0. (B) Bar graph shows the significant effect of the A2 protein in reducing the time to peak maximal absorbance in panel A. This represents 4 experiments using plasma from 2 healthy human donors (n = 4 paired participants).**P < .05 vs control (no A2), ***P < .007 vs control. OD, optical density.
The A2 protein modulates the rate of fibrin formation in vitro. (A) Fibrin was formed from using 10% healthy human plasma and vehicle control or the A2 protein (0-0.5 μM), and turbidity was measured at λ 405 nm. The tracings shown are the average of 4 separate experiments. The baseline for fibrin polymerization in different experimental groups is normalized to time 0. (B) Bar graph shows the significant effect of the A2 protein in reducing the time to peak maximal absorbance in panel A. This represents 4 experiments using plasma from 2 healthy human donors (n = 4 paired participants).**P < .05 vs control (no A2), ***P < .007 vs control. OD, optical density.
A2 protein alters and intercalates into the fibrin clot structure
Because the A2 protein accelerates the fibrin formation in plasma, we next examined the effect of the A2 protein on the polymerized fibrin structure using confocal microscopy. Figure 3A shows representative images of the fibrin structure formed in plasma from healthy human donors. Note that the 3-dimensional reconstruction of fibrin formed in the presence of the A2 protein (top right) gives the impression of greater density than the control clot (top left). Z-stack representative images show that the A2 protein apparently induced the formation of larger pores in the resultant dense-appearing clot (bottom right) in comparison with vehicle control (bottom left). In fact, as shown in Figure 3B, considerable variation in the network structure with the WT A2 protein (middle) was observed in comparison with plasma incubated with either vehicle control (left) or A2 mutant (right). Intrinsic differences in the resultant fibrin clot structure among donors were evident when we performed additional experiments using plasma from multiple healthy human donors. However, A2 protein–induced alteration in the fibrin structure was clearly distinguishable for each donor studied, and it significantly increased the porosity of the fibrin network (supplemental Figure 4A-B). We next evaluated the clot porosity by measuring flowthrough.27 The graph in supplemental Figure 4C gives the impression that the A2 protein apparently increased the porosity of the resultant clot (slightly higher flowthrough); however, the changes were not significant. The differences observed within the healthy donors and between the assays may be attributed to the content of other plasma proteins that participate in fibrin polymerization or fibrinogen,28,29 although the level of fibrinogen had minor effects on fibrin structure in vitro.30
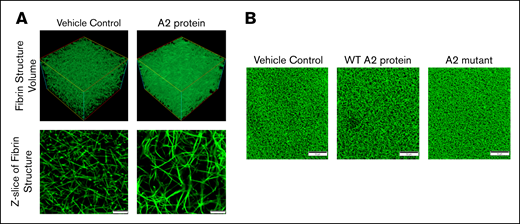
The effect of the A2 protein on the fibrin clot structure. (A) Confocal microscopy images of fibrin clots at a magnification of ×200 formed in plasma from healthy human donors. Note that the effect of the A2 protein (0.25 μM) on the resultant clot structure (right) is evident as compared with plasma incubated with vehicle control (left). Scale bars, 10 μM. (B) The WT A2 protein clearly altered the resultant fibrin structure as compared with plasma treated with vehicle control and the A2 mutant. Scale bars, 50 μM.
The effect of the A2 protein on the fibrin clot structure. (A) Confocal microscopy images of fibrin clots at a magnification of ×200 formed in plasma from healthy human donors. Note that the effect of the A2 protein (0.25 μM) on the resultant clot structure (right) is evident as compared with plasma incubated with vehicle control (left). Scale bars, 10 μM. (B) The WT A2 protein clearly altered the resultant fibrin structure as compared with plasma treated with vehicle control and the A2 mutant. Scale bars, 50 μM.
Fibrin formation in plasma or in the absence of blood cells differs from that in whole blood.31,32 Figure 4A shows images of fibrin clot structures, generated using healthy human whole blood in the presence of the A2 protein (0.5 μM). In comparison with whole blood mixed with vehicle control and dye only, it was evident that the A2 protein caused the formation of larger pores (top left) and specifically localized and overlapped with the fibrin network (top middle) and did not interact with other blood cells (top right vs bottom right, dye only). Additionally, confocal microscopy images of a higher magnification demonstrated the incorporation of the fluorescently labeled A2 protein directly into the fibrin clot network or fibrils (Figure 4B top middle), particularly at the location of fibrin branchings (white arrowheads). In contrast, the size of the pores formed in the presence of the A2 mutant (0.5 μM; Figure 4B bottom left) was smaller than that of those caused by the WT A2 protein (Figure 4B top left). In addition, the incorporation of the A2 mutant into the clot structure (Figure 4B bottom middle) was significantly lower (supplemental Figure 5) and did not form clusters in locations of fibrin branching as compared with the WT A2 protein (Figure 4B top middle). These outcomes indicate that the A2 protein is incorporated into the fibrin networks, and this interaction results in structural changes in the fibrin clot, causing larger pores.
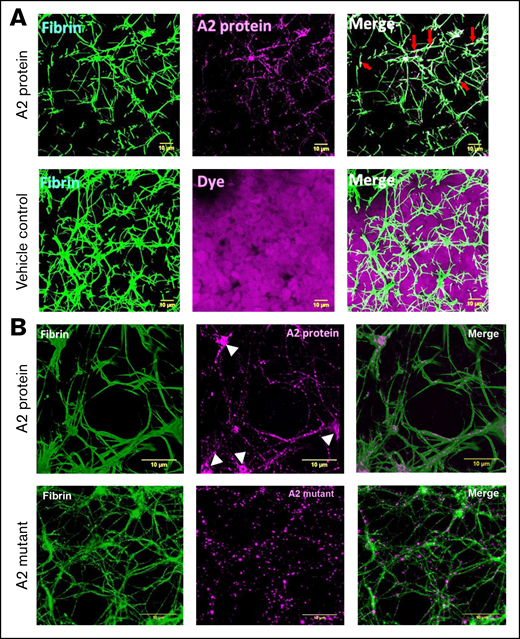
The A2 protein alters and is incorporated into the fibrin clot structure. (A) Representative of 3-dimensional confocal microscopy images of fibrin clots formed in whole blood from a healthy human donor mixed with either vehicle control or the A2 protein (0.5 μM) conjugated to Alexa Fluor 488 (purple). Fibrin was visualized by supplementing whole blood with 1% human fibrinogen conjugated to Alexa Fluor 647 (green). In comparison with the treatment of vehicle control, the A2 protein clearly modified the resultant fibrin structure. In addition, the A2 protein did not interact with the blood cells present in the mixture as compared with vehicle control containing dye only. Colocalization of fibrin and the A2 protein is viewed as a lighter green to white color in the right panel (red arrows). Scale bars, 10 μM. (B) A higher (300×) magnification demonstrates the incorporation of the A2 protein (top middle, purple) into the fibrin structure (top left, green). The white arrowheads point to the A2 protein bound to fibrin branching. In contrast, the A2 mutant did not increment the size of pores (bottom left, green) or formed clusters in fibrin branching (bottom middle, purple). Colocalization of fibrin and the A2 protein is viewed at 300× magnification (top right) as lighter magenta overlapping the fibrin fibers (as lighter green). In contrast, there is less colocalization or overlapping of the A2 mutant with the fibrin fibers (bottom right). Scale bars, 10 μM.
The A2 protein alters and is incorporated into the fibrin clot structure. (A) Representative of 3-dimensional confocal microscopy images of fibrin clots formed in whole blood from a healthy human donor mixed with either vehicle control or the A2 protein (0.5 μM) conjugated to Alexa Fluor 488 (purple). Fibrin was visualized by supplementing whole blood with 1% human fibrinogen conjugated to Alexa Fluor 647 (green). In comparison with the treatment of vehicle control, the A2 protein clearly modified the resultant fibrin structure. In addition, the A2 protein did not interact with the blood cells present in the mixture as compared with vehicle control containing dye only. Colocalization of fibrin and the A2 protein is viewed as a lighter green to white color in the right panel (red arrows). Scale bars, 10 μM. (B) A higher (300×) magnification demonstrates the incorporation of the A2 protein (top middle, purple) into the fibrin structure (top left, green). The white arrowheads point to the A2 protein bound to fibrin branching. In contrast, the A2 mutant did not increment the size of pores (bottom left, green) or formed clusters in fibrin branching (bottom middle, purple). Colocalization of fibrin and the A2 protein is viewed at 300× magnification (top right) as lighter magenta overlapping the fibrin fibers (as lighter green). In contrast, there is less colocalization or overlapping of the A2 mutant with the fibrin fibers (bottom right). Scale bars, 10 μM.
Because the fibrin network is susceptible to cleavage by plasmin, we examined the hypothesis that the A2 protein binding to the fibrin network structure influences fibrin degradation by plasmin. The fibrin polymerization and fibrinolysis assays were conducted in healthy human plasma mixed with tissue plasminogen activator in the presence of either vehicle control or the A2 protein. Although the A2 protein reduced the time to reach maximum absorbance, it did not have a marked effect on fibrin degradation, as compared with plasma incubated with vehicle control (supplemental Figure 6). These outcomes indicate that the A2 protein specifically influences the rate of fibrin polymerization and its resultant clot structure without altering fibrin degradation by plasmin in healthy plasma in vitro.
A2 protein–fibrin interaction attenuates microvascular thrombosis in vivo
We next investigated whether the ability of the A2 protein to engage fibrin and induce structural changes in vitro translates to attenuation of microvascular thrombosis in an endotoxemic in vivo mouse model. As previously described,13 we treated the LPS-challenged mice with either saline, the WT A2 protein, or the A2 mutant (4.0 mg/kg), with diminished fibrin binding activity as negative control, 1.5 hours after the LPS insult. Figure 5A shows that survival of LPS-challenged mice did improve with the WT A2 protein as compared with saline- or A2 mutant–treated mice. Moreover, in comparison with sick animals treated with saline or A2 mutant, the WT A2 protein was more effective in diminishing fibrin deposition in glomeruli, as shown in Figure 5B-C.
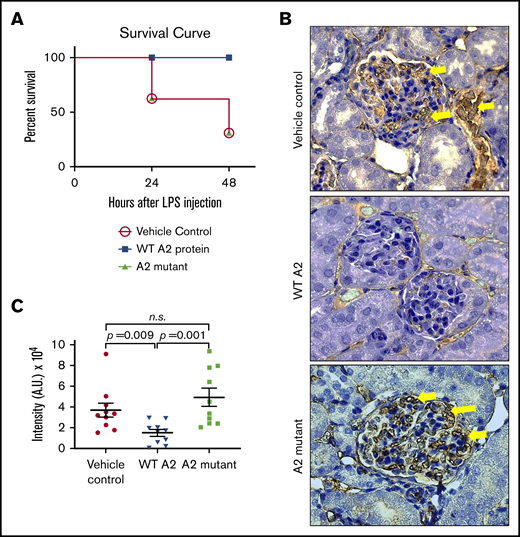
The A2 protein exerts its beneficial effect via fibrin in vivo. (A) Survival curve depicting the effect of the A2 mutant (n = 12) in LPS-treated mice as compared with LPS-treated mice with either the WT A2 protein (n = 10) or saline (n = 6). The difference is statistically significant (P < .05). (B) Kidneys were harvested at 24 hours after the administration of LPS to mice and stained for fibrin. Dark brown color depicts the fibrin deposition (yellow arrow), and randomized areas were selected for analysis using ImageJ. (C) In comparison with mice treated with the WT A2 protein, an increased fibrin deposition was notable for mice that received saline or the A2 mutant (n = 10 per group, unpaired subjects). The difference between the WT A2 and control or the A2 mutant protein was significant. A.U., arbitrary unit.
The A2 protein exerts its beneficial effect via fibrin in vivo. (A) Survival curve depicting the effect of the A2 mutant (n = 12) in LPS-treated mice as compared with LPS-treated mice with either the WT A2 protein (n = 10) or saline (n = 6). The difference is statistically significant (P < .05). (B) Kidneys were harvested at 24 hours after the administration of LPS to mice and stained for fibrin. Dark brown color depicts the fibrin deposition (yellow arrow), and randomized areas were selected for analysis using ImageJ. (C) In comparison with mice treated with the WT A2 protein, an increased fibrin deposition was notable for mice that received saline or the A2 mutant (n = 10 per group, unpaired subjects). The difference between the WT A2 and control or the A2 mutant protein was significant. A.U., arbitrary unit.
Ex vivo assays demonstrated an increased rate of fibrin formation in plasma from endotoxemic mice treated with the A2 protein
We next analyzed the fibrin clot structure in plasma of endotoxemic mice. Because the A2 protein is found in circulation 2 hours after its injection in mice,13 we obtained plasma from LPS-treated mice 2 hours after treatment with saline (control) or A2 variants. The fibrin clot structure observed in plasma from a sick mouse that received the WT A2 protein was significantly different (larger pores) than that of saline or A2 mutant LPS-challenged mice (Figure 6A-B). These outcomes indicate that the engagement of the A2 protein with fibrin seems to be an important mechanism to attenuate microvascular thrombosis in our LPS-induced DIC murine model.
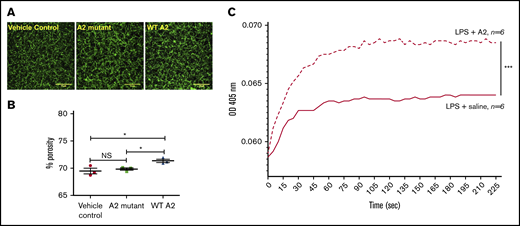
The A2 protein in endotoxemic mice modulated fibrin formation ex vivo. (A-B) Confocal images of the fibrin structure in endotoxemic plasma collected at 2 hours after the injection of the A2 variants or vehicle control were analyzed using ImageJ. (A) Notably, larger pores were observed in the resultant fibrin clot in plasma from mice treated with the WT A2 protein (right) in comparison with mice treated with vehicle control or the A2 mutant (left or middle, respectively; n = 3 unpaired subjects). Representative of 3 experiments using 3 different mice per group. Scale bars, 10 μM. (C) Plasma samples from LPS-treated mice were obtained at 2 hours after IP injection of A2 protein (4.0 mg/kg) or saline. Fibrin polymerization was measured by turbidity at 405 nm. Turbidity curves represent the average of 6 separate experiments for each condition tested: with A2 protein or saline. *P = .036 vs control or mutant, ***P < .0001. NS, not significant.
The A2 protein in endotoxemic mice modulated fibrin formation ex vivo. (A-B) Confocal images of the fibrin structure in endotoxemic plasma collected at 2 hours after the injection of the A2 variants or vehicle control were analyzed using ImageJ. (A) Notably, larger pores were observed in the resultant fibrin clot in plasma from mice treated with the WT A2 protein (right) in comparison with mice treated with vehicle control or the A2 mutant (left or middle, respectively; n = 3 unpaired subjects). Representative of 3 experiments using 3 different mice per group. Scale bars, 10 μM. (C) Plasma samples from LPS-treated mice were obtained at 2 hours after IP injection of A2 protein (4.0 mg/kg) or saline. Fibrin polymerization was measured by turbidity at 405 nm. Turbidity curves represent the average of 6 separate experiments for each condition tested: with A2 protein or saline. *P = .036 vs control or mutant, ***P < .0001. NS, not significant.
Next, we examined if the A2 protein influences fibrin polymerization ex vivo using plasma from mice with (LPS) endotoxemia. Blood was drawn from the LPS-challenged mice 2 hours after the IP injection of either saline or the A2 protein (4.0 mg/kg). Plasma was obtained, and as described earlier, we used the turbidity assay to assess fibrin polymerization. In comparison with mice with LPS and saline, fibrin formation was significantly potentiated in plasma from sick mice injected with the A2 protein, as shown in Figure 6C. Moreover, the presence of the A2 protein markedly enhanced the rate of change (determined as above) in comparison with animals that received saline (slope, 29.7 × 10−5 vs 16.6 × 10−5, respectively). However, the time to reach maximum turbidity was similar in both groups (∼95 seconds). Thus, the A2 protein increased the rate of fibrin polymerization in human blood in vitro (Figure 2) and in endotoxemic murine blood ex vivo. These results suggest that the beneficial effect of the A2 protein in our mouse model for DIC is dependent on its ability to engage fibrin.
A2 protein does not interfere with experimental thrombosis
Several clinical trials have been conducted to attenuate the morbidity and mortality associated with sepsis-associated DIC without success, because the tested antithrombotic drugs can cause severe bleeding adverse effects.33 Because the A2 protein did not affect the tail bleeding time in mice,13 we further examined the effect of the A2 protein in a murine thrombosis model by using intravital microscopy. At the dose tested in our LPS model, the A2 protein did not alter thrombus formation in vivo as compared with animals treated with control vehicle only (supplemental Figure 7A). In parallel, the A2 protein added to whole blood from healthy human donors did not have a profound effect on platelet adhesion or thrombus formation on collagen-coated surfaces at high shear rates in vitro (supplemental Figure 7B). These results imply that the A2 protein does not impair experimental thrombosis.
Discussion
Previously, we reported the effectiveness of the recombinant A2 domain of VWF, the A2 protein, in attenuating microvascular thrombosis and improving survival in a murine model for LPS-induced DIC.13 Although we described the binding of the A2 protein to fibrin, the precise mechanism by which the A2 protein exerted its beneficial effect in vivo remained elusive. This is because besides fibrin, the A2 protein can bind to both the A1 domain of VWF and vimentin, 2 additional ligands that are involved in platelet adhesion and thrombus formation.18,19,34 In this study, we have examined the hypothesis that fibrin is the target for the A2 protein during systemic inflammation. Comparative analyses between the WT A2 protein and A2 (E1567A) mutant, which selectively inhibited the binding to fibrin but retained WT-binding activity for both the A1 domain and vimentin, validated that the A2 protein functions through the interaction with fibrin in vivo.
The A2 protein does not act as an anticoagulant, but rather, it directly increases the rate of fibrin polymerization and is incorporated into the formed fibrin network, causing larger pores in the fibrin network formed in plasma from different healthy human donors. Consistent with studies in healthy human plasma, the fibrin clot structure formed ex vivo in plasma derived from endotoxemic mice treated with the A2 protein also demonstrated increased porosity, compared with corresponding sick animals with mutant A2. It has been reported that changes in the architecture of the fibrin clot are associated with a high risk for thrombosis in certain diseases,7,9,10,35-37 and several studies have indicated that an increase in fibrin network porosity facilitates fibrinolysis.38-40 Such a potential mechanistic effect of the A2 protein is consistent with the observation that microvascular thrombosis as well as widespread fibrin deposition in kidneys was markedly reduced in the A2-treated endotoxemic sick mice. These outcomes and the increase in porosity within the fibrin network suggest that the A2 protein could also influence fibrinolysis. However, the absence of a significant effect of the A2 protein on fibrinolysis in the in vitro assays using healthy human plasma (supplemental Figure 5) and the inconsistent results in the fibrinolysis assays using plasma from endotoxemic mice (not shown) preclude us from suggesting that the A2 protein may modulate fibrinolysis. Therefore, more studies are necessary to elucidate why the A2 protein effectively reduced fibrin deposition in an in vivo murine model for LPS-induced DIC, while altering fibrin polymerization but apparently not the fibrinolysis in ex vivo studies.
It has been published that fibrin thickness is dependent on a variety of parameters, including the rates of fibrinopeptide A cleavage, protofibril formation, and fiber initiation.41 On the basis of the profiles in Figure 2A, it can be argued that the action of the A2 protein occurs during the first step in fibrin formation, possibly altering the rate of fibrinopeptide A removal or lag phase, which was affected by the A2 protein at 2 different concentrations. Additionally, the effects of the A2 protein on the observed turbidity profiles and initial rates of change (slope) are consistent with A2 altering the rate of fiber growth as previously defined.41 Thus, because the A2 protein preferably binds to fibrin monomer,13 it is possible that the bound A2 protein increments the interactions between fibrin monomers that lead to the formation of protofibrils, thereby affecting the thickness and architecture of the resultant fibrin clot structure.42 Note that the effect described in this study for the A2 protein using human plasma distinctly contrasts our previous report, in which the A2 protein delayed fibrin polymerization in a system containing only purified fibrinogen.13 Future studies will be needed to address the underpinning mechanisms by which the A2 protein directly affects the structural features of the resultant fibrin network structure, particularly under systemic inflammation. Nevertheless, our studies in human and mice plasma provide evidence that the A2 protein accelerates fibrin polymerization and engages and interacts with the fibrin network to augment its porosity. Importantly, these structural changes in the fibrin network correlate with attenuated microvascular thrombosis in vivo.
The A2 protein was effective in diminishing fibrin deposition and fibrin-rich microthrombi formation (hallmarks of DIC) in the kidneys of mice with endotoxemia. To date, acute DIC is frequently managed with anticoagulants, but the use of these therapies increases the risk of bleeding; therefore, the ideal treatment for DIC should aim at reducing both the severity of bleeding and/or thrombosis without affecting hemostasis. As we described, the A2 protein did not alter tail bleeding time in mice13 or provoke excessive bleeding in endotoxemic mice, and it did not affect the occlusion time in a mouse model of thrombosis. These data provide evidence that the A2 protein could attenuate DIC associated with systemic inflammation, sepsis, or other conditions without causing increasing bleeding.
Another fascinating and clinically relevant result obtained from this study is the protective effect conferred by the A2 protein in attenuating both microvascular thrombosis and fibrin deposition. Here we demonstrated the efficacy of the A2 protein but not the mutant A2 protein, when administrated 1.5 hours after the endotoxin insult in our LPS-induced DIC murine model.13 Thus, the A2 protein demonstrated its pharmacological efficacy when the animals were already sick, modeling the typical presentation of systemic inflammation in patients who seek medical attention after the development of symptoms.
Normally, the A2 domain is buried within the globular morphology of the plasma VWF. However, this A2 domain can be found exposed when plasma VWF is unfolded by the influence of high hydrodynamic forces43 and in newly released VWF molecules from the stimulated endothelium.44 Previously, we reported the effectiveness of the A2 protein in blocking the interaction of full-length VWF with fibrin.13 However, the effects of the purified recombinant A2 protein in fibrin formation reported in this study do not necessarily reveal novel biological roles for the A2 domain in the context of intact full-length VWF. In addition, the A2 protein does not contain the posttranslational modifications of the native A2 domain in VWF.45 Nevertheless, those novel concepts/mechanisms are also being investigated.
In summary, the A2 protein binds to fibrin and accelerates fibrin polymerization, altering the physical structure of the resultant polymerized fibrin clot. The A2 protein diminished fibrin deposition and microvascular thrombosis in kidneys in an animal model for systemic inflammation by targeting fibrin. Therefore, the A2 protein is a novel therapeutic approach in patients with uninhibited activated coagulation and disseminated fibrin deposition, as in DIC. Additional large animal and human studies are warranted to confirm our findings and further develop the A2 protein as a therapeutic agent.
Data sharing requests can be e-mailed to the corresponding author, Miguel A. Cruz (miguelc@bcm.edu).
Acknowledgments
The authors thank Kimberly Langlois for excellent technical assistance.
This work was supported by National Institutes of Health (NIH), National Institute of General Medical Sciences grant R01 GM112806 (M.A.C., T.C.N., and K.V.V.); NIH, National Institute of Neurological Disorders and Stroke grant R01 NS094280 (M.A.C.); Merit Review Award I01 BX002551 from the US Department of Veterans Affairs Biomedical Laboratory Research and Development Service (R.E.R.); NIH, National Heart, Lung, and Blood Institute grant T32 HL139425 (M.M.-V.); the Alkek Foundation; the Fondren Foundation; and the Mary Gibson Foundation.
The content of this publication does not represent the views of the Department of Veterans Affairs or the United States Government.
Authorship
Contribution: C.V. and M.M.-V. designed and performed experiments, analyzed data, and helped write the manuscript; R.E.R. performed experiments and analyzed data; K.V.V. helped write the manuscript; N.S., C.B., T.P., A.T., M.A., and F.L. performed experiments; and T.C.N. and M.A.C. designed and performed experiments, analyzed and interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: M.A.C. is the founder and CSO of A2 Therapeutics, Inc. The remaining authors declare no competing financial interests.
Correspondence: Miguel A. Cruz, Baylor College of Medicine/Center for Translational Research on Inflammatory Diseases, Michael E. DeBakey Veterans Affairs Medical Center, Medicine/Section of Cardiovascular Research, 2002 Holcombe Blvd, B-109, Room 146, Houston, TX 77030; e-mail: miguelc@bcm.edu.

