

Key Points
Downregulation of the IL-6/STAT3 pathway in CD4+FOXP3− T cells correlates with better outcome of HR-MDS patients treated with azacitidine.
The AZA-mediated downregulation of IL-6/STAT3 axis reestablishes the STAT signaling architecture of CD4+ T cells at the single cell level.

Abstract
CD4+ T cells orchestrate immune responses and are actively engaged in shaping tumor immunity. Signal transducer and activator of transcription (STAT) signaling controls the epigenetic tuning of CD4+ T-cell differentiation and polarization, and perturbed STAT signaling networks in CD4+ T cells subvert antitumor immunity in malignancies. Azacitidine (AZA), the mainstay therapy for high-risk myelodysplastic syndromes (HR-MDS), affects CD4+ T-cell polarization and function, but whether this contributes to AZA efficacy is currently unknown. By using functional proteomic, transcriptomic, and mutational analyses in 73 HR-MDS patients undergoing AZA therapy, we demonstrate that responding patients exhibited a coordinated CD4+ T-cell immune response and downregulated the inflammatory cytokine signaling pathways in CD4+ T cells after AZA, in contrast to nonresponders who upregulated the same pathways. We further observed an AZA-mediated downregulation of intereukin-6 (IL-6)—induced STAT3 phosphorylation in CD4+FOXP3− conventional T cells (Tcons) that correlated independently with better response and survival, whereas it was also not associated with the mutation number and profile of the patients. The AZA-induced downregulation of IL-6/STAT3 axis in Tcons restored the STAT signaling architecture in CD4+ T-cell subsets, whereas STAT signaling networks remained disorganized in patients who upregulated IL-6/STAT3 activity in Tcons. Given the pivotal role of CD4+ T cells in adaptive immunity, our findings suggest that the downregulation of the IL-6/STAT3 pathway in Tcons potentially constitutes a previously unrecognized immune-mediated mechanism of action of AZA and sets the scene for developing rational strategies of AZA combinations with IL-6/STAT3 axis inhibitors.
Introduction
CD4+ T cells hold a cardinal position in orchestrating both normal and antitumor immune responses. Beyond their central role in coordinating antibody and CD8+ cell-mediated anticancer responses,1 effector CD4+ T cells can mediate direct cytotoxicity through cytolytic mechanisms,2-4 whereas CD4+ chimeric antigen receptor T cells show identical killing efficacy with their CD8+ counterparts.5
Myelodysplastic syndromes (MDS) comprise a heterogeneous group of myeloid malignancies characterized by ineffective hematopoiesis and variable rates of leukemic transformation.6 Disease progression in MDS is accompanied by a declining immune competence7 and expansion of FOXP3+ regulatory T cells (Tregs) and T-helper 17 (Th17) cells that retain their suppressive function and migratory capacity.8-10 However, the role of the other CD4+ T-cell subsets in the evolutionary trajectory of MDS and the immune evasion of the leukemic clone is uncertain. Azacitidine (AZA) is the only agent to prolong survival in higher-risk MDS (HR-MDS) patients 11 and remains the mainstay of treatment 15 years after its approval. In vitro and in vivo studies indicate that AZA inhibits CD4+ T-cell proliferation with simultaneous induction of Tregs,12,13 but findings in HR-MDS patients are conflicting, showing mixed effects on CD4+ T-cell subsets.14-16
The interplay between DNA methylation and signal transducer and activator of transcription (STAT) signaling is essential for the epigenetic control of differentiation and polarization of CD4+ cells.17 Defective immune signaling via STATs leads to compromised CD4+ T cell–mediated antitumor responses and propel immune evasion in various malignancies.18-20 We have previously shown that AZA therapy directly affects signal transduction pathways and can partially restore the pathologic STAT3/5 biosignature of the clonal CD34+ progenitors in responding patients.21 However, the STAT signaling alterations in CD4+ T cells of HR-MDS patients and the effect of AZA on immune signaling are currently unknown.
By combining functional proteomic, transcriptional, and mutational analyses, we studied the immune architecture and the STAT signaling profile of CD4+ T cells and their associations with clinical and molecular data in HR-MDS patients undergoing AZA therapy. We demonstrate that downregulation of the interleukin-6 (IL-6)–mediated STAT3 phosphorylation in CD4+FOXP3− conventional T cells (Tcons) restores the connectivity of STAT signaling networks in CD4+ T-cell subsets and can act as a predictor of better response and longer overall survival (OS).
Patients and methods
Patients
Samples were collected from 73 patients before AZA administration, at day 15 of the first cycle, and at day 28 from the beginning of the sixth cycle (supplemental Table 1). Peripheral blood (PBMCs) and bone marrow mononuclear cells (BMMCs) were isolated using density gradient centrifugation. Patients were treated with AZA in a nonclinical trial setting at an initial dose of 75 mg/m2 subcutaneously for 7 days on 28-day cycles. Dose reductions up to 50% of the initial dose and/or treatment delays were considered for myelotoxicity or myelosuppression-related complications. Granulocyte colony-stimulating factors were used at the discretion of the treating physician, whereas no erythropoiesis stimulating agents were administered to patients. Response to therapy was evaluated using the International Working Group Response Criteria for MDS.22 Heavily transfused patients were defined as those requiring ≥4 red blood cell units for 8 weeks.23 All samples were collected following institutional review board approval, and written informed consent was obtained from all patients in accordance to the Declaration of Helsinki.
Antibodies and data acquisition
The following monoclonal antibodies were used in various multiparametric panels: CD3 (SK7), CD4 (SK3), CD8 (SK1), CD56 (NCAM16.2), CD45 (2D1), INFγ/IL4 (25723.11 3010.211), IL17a (SCPL1362), anti-Perforin (δ G9), CD126 (IL6Ra) (UV4), gp130 (AM64), pSTAT1 (pY701), pSTAT3 (pY705), pSTAT5 (pY694), and PD1 (EH12.1) from BD Biosciences and CD27 (M-T271), CD45RA (HI100), Helios (22F6), and FOXP3 (206D) from Biolegend. Fluorescence minus one (FMO) was used as a negative control. Data were acquired on a 6-color fluorescence-activated cell sorting (FACS) Canto flow cytometer (BD Biosciences) and were analyzed by FlowJo (Treestar).
Immune profiling by multiparametric flow cytometry
PBMCs were stained with markers specific for differentiation, polarization, and functional state of CD4+ and CD8+ T lymphocytes. Naïve cells were defined as CD27+CD45RA+, terminal effector memory cells as CD27−CD45RA+, effector memory as CD27−CD45RA−, and central memory cells as CD27+CD45RA−. PD-1 was used as an exhaustion marker, and the cytotoxic capacity was measured via Perforin levels. The T-helper polarization status was assessed by intracellular cytokine staining as described elsewhere.10
Transcriptional profiling by RNA-seq
Analysis of RNA-seq data from CD4+ bone marrow T cells deposited at the European Molecular Biology Laboratory–European Bioinformatics Insitute (EMBL-EBI) repository (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-8208 was performed as described in the supplemental Appendix.
Real-time quantitative polymerase chain reaction
CD4+ T cells from 4 responder and 4 nonresponder patients were isolated using the CD4+ T-Cell Isolation Kit by Miltanyi Biotec according to the manufacturer’s instructions before AZA administration and at day 28 from the beginning of the sixth cycle. Extraction of mRNA was performed in isolated CD4+ cells (purity ≥98%) using the RNeasy Mini Kit (Qiagen) and reverse transcription using the QuantiNova Reverse Transcription Kit (Qiagen). Quantification of STAT3 target genes was performed using Sybr Green (Qiagen) in a Chromo4TM Real-Time Detector (Bio-Rad, Hercules, CA), using gene-specific primers (supplemental Table 2). Data acquisition and analysis were performed in Opticon Monitor 3. Gene expression was calculated using the 2−ΔΔCt method and normalized against GAPDH.
Single-cell phospho-specific flow cytometry in CD4+ T cells
PBMCs were processed as previously described.21 Cells were stimulated for 15 minutes with human recombinant IL-2 (100 ng/mL, Miltenyi Biotec), IL-6 (100 ng/mL, Miltenyi Biotec), or interferon a2a (IFNα) (10,000 U/mL, Miltenyi Biotec). Cell fixation was performed with FIX/PERM FOXP3 Fixation buffer (Biolegend) and permeabilization with Perm III (BD Biosciences). Basal phosphorylation levels were expressed as the log2 ratio of median fluorescence intensity (MFI) of unstimulated phospho-STATs divided by the FMO control, namely log2[MFI (unstimulated)/MFI (FMO)] and potentiated levels as log2[MFI (stimulated)/MFI (unstimulated)]. The following nodes (ie, combinations of stimuli/target) were studied: IL-2/pSTAT5, IL-6/pSTAT1, IL-6/pSTAT3, IFNα/pSTAT1, and IFNα/pSTAT5. To assess the up- or downregulation of each single node after AZA therapy, we used the following equation: [(MFI stimulated at sixth cycle/MFI unstimulated at sixth cycle) − (pretreatment MFI stimulated/pretreatment MFI unstimulated)]/(pretreatment MFI stimulated/pretreatment MFI unstimulated).
Immunoassays
Quantification of IL-6 levels in isolated EDTA plasma from bone marrow aspirates was performed by engaging the V-plex Proinflammatory Panel 1 (Human) Kit (Meso Scale Discovery), using a Meso QuickPlex SQ 120 instrument (Meso Scale Discovery), according to the manufacturer’s instructions. The levels of soluble IL-6RA were measured using the Human IL-6R α Quantikine ELISA Kit (R&D Systems), according to the manufacturer’s instructions.
Next-generation sequencing
DNA was extracted from BMMCs or PBMCs collected before AZA administration and at day 28 from the beginning of the sixth cycle. Next-generation sequencing was performed by either an in-house targeted panel or using the Illumina Trusight Myeloid Panel (supplemental Appendix).
Correlation network analysis and connectivity analysis
The frequency of each immune cell subset per patient was calculated by flow cytometry. Pairwise Spearman correlations were calculated for each immune cell subset, and hierarchical clustering was performed to arrange the correlation matrix. Connectivity analysis was assessed by creation of an adjacency matrix from the correlation matrix of responders to AZA using a Spearman correlation coefficient of 0.4 as the threshold. The graph of the adjacency matrix was created by aMatReader application of Cytoscape software and visualizes all positive correlations present in the adjacency matrix for each subset.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad Inc, La Jolla, CA) and IBM SPSS Statistics 23. Comparisons were performed using χ2, Fisher’s exact, Mann-Whitney, Kruskal-Wallis, Wilcoxon signed rank, and Friedman tests as appropriate. Survival analysis was performed by Kaplan-Meier and log-rank test. OS was defined as the time from AZA initiation to death from any cause and event-free survival (EFS) as the time from AZA initiation to disease progression, relapse, or death. Surviving patients were censored at last follow-up. Significance was set at P < .05.
Results
Responders to AZA demonstrate a coordinated CD4+ T-cell immune response
Patient characteristics are summarized at Table 1. Responders to AZA were defined as those who achieved complete (CR, 29.8%) or partial (3%) remission and hematologic improvement (9.6%), whereas nonresponders were defined as those with stable disease without hematologic improvement (23.3%) and failure to AZA (34.3%).
Baseline patient characteristics and clinical information
| Patients (n = 73) | IL-6/STAT3_Down (n = 28) | IL-6/STAT3_Up (n = 26) | P | |
|---|---|---|---|---|
| Age (median, range), y | 72.9 (32.78-86.7) | 73.1 (48.6-86.7) | 71.05 (34.6-83) | .634 |
| >65 | 58 (79.4%) | |||
| <65 | 15 (20.6%) | |||
| Sex | .747 | |||
| Male | 56 (76.7%) | 21 (75%) | 21 (80.8%) | |
| Female | 17 (23.2%) | 7 (25%) | 5 (19.2%) | |
| Baseline blood counts | ||||
| Hemoglobin, g/dL | 9.1 (3-13.9) | 9.1 (6.2-12.1) | 9.35 (6.9-13.9) | .473 |
| ANC, ×109/L | 1.58 (0.04-17.76) | 1.69 (0.04-16.39) | 1.75 (0.25-17.76) | .489 |
| Platelets, ×109/L | 77.5 (5-490) | 80 (5-481) | 82 (15-301) | .482 |
| Number of completed cycles | .007 | |||
| Median (range) | 7 (2-36) | 15 (6-34) | 6 (6-35) | |
| WHO classification | .624 | |||
| RCMD | 7 (9.5%) | 2 (7.2%) | 4 (15.4%) | |
| RAEB-II | 38 (52%) | 14 (50%) | 9 (34.6%) | |
| CMML-II | 15 (20.5%) | 6 (21.4%) | 7 (26.9%) | |
| AML-MDS | 13 (18%) | 6 (21.4%) | 6 (23.1%) | |
| IPSS | .901 | |||
| Intemediate-1 | 12 (16.4%) | 4 (14.3%) | 4 (15.4%) | |
| Intermediate-2 | 30 (41.1%) | 13 (46.4%) | 12 (46.1%) | |
| High | 26 (35.6%) | 11 (39.3%) | 8 (30.8%) | |
| N/A | 5 (6.9%) | — | 2 (7.7%) | |
| WPSS | .542 | |||
| Intermediate | 1 (1.4%) | — | — | |
| High | 34 (46.6%) | 13 (46.4%) | 11 (42.3%) | |
| Very high | 17 (23.2%) | 9 (32.2%) | 5 (19.2%) | |
| N/A | 21 (28.8%) | 6 (21.4%) | 10 (38.5%) | |
| IPSS-R | .733 | |||
| Intermediate | 5 (6.9%) | 1 (3.6%) | 2 (7.7%) | |
| High | 33 (45.2%) | 16 (57.1%) | 12 (46.1%) | |
| Very high | 28 (38.3%) | 11 (39.3%) | 9 (34.6%) | |
| N/A | 7 (9.6%) | — | 3 (11.6%) | |
| IPSS-R cytogenetic risk | .858 | |||
| Good | 33 (45.2%) | 11 (39.3%) | 12 (46.1%) | |
| Intermediated | 12 (16.4%) | 6 (21.4%) | 5 (19.2%) | |
| Poor | 17 (23.2%) | 9 (32.2%) | 6 (23.1%) | |
| Very poor | 6 (8.3%) | 2 (7.1%) | 1 (3.9%) | |
| N/A | 5 (6.9%) | — | 2 (7.7%) | |
| PB blasts | .213 | |||
| Present | 37 (50.7%) | 16 (57.1%) | 13 (50%) | |
| Absent | 26 (35.6%) | 5 (17.9%) | 10 (28.5%) | |
| N/A | 10 (13.7%) | 7 (25%) | 3 (11.5%) | |
| BM blasts | .074 | |||
| >15% | 35 (48%) | 5 (17.9%) | 11 (42.3%) | |
| ≤15% | 38 (52%) | 23 (82.1%) | 15 (57.7%) | |
| Transfusions ≥4 RBC U/8 wk | .99 | |||
| Yes | 23 (31.5%) | 6 (21.4%) | 6 (23.1%) | |
| No | 50 (68.5%) | 22 (78.6%) | 20 (76.9%) | |
| Best response | .001 | |||
| Complete and partial remission | 24 (31.3%) | 18 (64.3%) | 4 (15.4%) | |
| Hematologic improvement | 7 (10.5%) | 2 (7.1%) | 5 (19.2%) | |
| Stable disease | 17 (25.4%) | 4 (14.3%) | 6 (23.1%) | |
| Failure | 25 (32.8%) | 4 (14.3%) | 11 (42.3%) |
| Patients (n = 73) | IL-6/STAT3_Down (n = 28) | IL-6/STAT3_Up (n = 26) | P | |
|---|---|---|---|---|
| Age (median, range), y | 72.9 (32.78-86.7) | 73.1 (48.6-86.7) | 71.05 (34.6-83) | .634 |
| >65 | 58 (79.4%) | |||
| <65 | 15 (20.6%) | |||
| Sex | .747 | |||
| Male | 56 (76.7%) | 21 (75%) | 21 (80.8%) | |
| Female | 17 (23.2%) | 7 (25%) | 5 (19.2%) | |
| Baseline blood counts | ||||
| Hemoglobin, g/dL | 9.1 (3-13.9) | 9.1 (6.2-12.1) | 9.35 (6.9-13.9) | .473 |
| ANC, ×109/L | 1.58 (0.04-17.76) | 1.69 (0.04-16.39) | 1.75 (0.25-17.76) | .489 |
| Platelets, ×109/L | 77.5 (5-490) | 80 (5-481) | 82 (15-301) | .482 |
| Number of completed cycles | .007 | |||
| Median (range) | 7 (2-36) | 15 (6-34) | 6 (6-35) | |
| WHO classification | .624 | |||
| RCMD | 7 (9.5%) | 2 (7.2%) | 4 (15.4%) | |
| RAEB-II | 38 (52%) | 14 (50%) | 9 (34.6%) | |
| CMML-II | 15 (20.5%) | 6 (21.4%) | 7 (26.9%) | |
| AML-MDS | 13 (18%) | 6 (21.4%) | 6 (23.1%) | |
| IPSS | .901 | |||
| Intemediate-1 | 12 (16.4%) | 4 (14.3%) | 4 (15.4%) | |
| Intermediate-2 | 30 (41.1%) | 13 (46.4%) | 12 (46.1%) | |
| High | 26 (35.6%) | 11 (39.3%) | 8 (30.8%) | |
| N/A | 5 (6.9%) | — | 2 (7.7%) | |
| WPSS | .542 | |||
| Intermediate | 1 (1.4%) | — | — | |
| High | 34 (46.6%) | 13 (46.4%) | 11 (42.3%) | |
| Very high | 17 (23.2%) | 9 (32.2%) | 5 (19.2%) | |
| N/A | 21 (28.8%) | 6 (21.4%) | 10 (38.5%) | |
| IPSS-R | .733 | |||
| Intermediate | 5 (6.9%) | 1 (3.6%) | 2 (7.7%) | |
| High | 33 (45.2%) | 16 (57.1%) | 12 (46.1%) | |
| Very high | 28 (38.3%) | 11 (39.3%) | 9 (34.6%) | |
| N/A | 7 (9.6%) | — | 3 (11.6%) | |
| IPSS-R cytogenetic risk | .858 | |||
| Good | 33 (45.2%) | 11 (39.3%) | 12 (46.1%) | |
| Intermediated | 12 (16.4%) | 6 (21.4%) | 5 (19.2%) | |
| Poor | 17 (23.2%) | 9 (32.2%) | 6 (23.1%) | |
| Very poor | 6 (8.3%) | 2 (7.1%) | 1 (3.9%) | |
| N/A | 5 (6.9%) | — | 2 (7.7%) | |
| PB blasts | .213 | |||
| Present | 37 (50.7%) | 16 (57.1%) | 13 (50%) | |
| Absent | 26 (35.6%) | 5 (17.9%) | 10 (28.5%) | |
| N/A | 10 (13.7%) | 7 (25%) | 3 (11.5%) | |
| BM blasts | .074 | |||
| >15% | 35 (48%) | 5 (17.9%) | 11 (42.3%) | |
| ≤15% | 38 (52%) | 23 (82.1%) | 15 (57.7%) | |
| Transfusions ≥4 RBC U/8 wk | .99 | |||
| Yes | 23 (31.5%) | 6 (21.4%) | 6 (23.1%) | |
| No | 50 (68.5%) | 22 (78.6%) | 20 (76.9%) | |
| Best response | .001 | |||
| Complete and partial remission | 24 (31.3%) | 18 (64.3%) | 4 (15.4%) | |
| Hematologic improvement | 7 (10.5%) | 2 (7.1%) | 5 (19.2%) | |
| Stable disease | 17 (25.4%) | 4 (14.3%) | 6 (23.1%) | |
| Failure | 25 (32.8%) | 4 (14.3%) | 11 (42.3%) |
IL-6/STAT3_Down represents IL-6/STAT3 downregulators and IL-6/STAT3_Up represents IL-6/STAT3 upregulators.
ANC, absolute neutrophil count; BM, bone marrow; CMML-II, chronic myelomonocytic leukemia type 2; IPSS-R, Revised International Prognostic Scoring System; N/A, not applicable/not available; PB, peripheral blood; RAEB-II, refractory anemia with excess blasts type 2; RBC, red blood cell; RCMD, refractory cytopenia with multilineage dysplasia; WHO, World Health Organization; WPSS, WHO classification-based prognostic scoring system.
To address the role of CD4+ T-cell subsets in the immune response at a systems level, we initially performed network analysis of immune populations before treatment initiation. For this, we calculated for each patient the frequencies of peripheral blood natural killer (NK)- and T-cell subsets, bone marrow and peripheral blood blasts, and absolute neutrophil counts and then performed pairwise correlations across responders and nonresponders to AZA. We observed several coordinated modules indicative of a highly organized response in responders to AZA, whereas no module was formed in nonresponders (Figure 1A-B). However, pretreatment measurements of CD4+ T-cell subsets showed no numerical differences between responders and nonresponders to AZA (Figure 1C). We further assessed the degree of connectivity of each subset by generating a rank-ordered adjacency matrix from the correlation data of responding patients. Three of the top 4 ranking subpopulations were CD4+ in origin (Figure 1D). The other subset was central memory CD8+ T cells, consistent with their higher antitumor activity.24 These findings indicate the existence of coordinated cellular immunity only in patients responding to AZA and point to CD4+ cells as a pivotal orchestrator of this immune response.

CD4+T-cell subsets mediate coordination of the immune response in HR-MDS patients responding to AZA. (A-B) Pairwise correlations and hierarchical clustering of pretreatment peripheral blood (PB) and bone marrow (BM) blasts, absolute neutrophil count, and T-, and NK-cell frequencies in responders (n = 27) (A) and nonresponders (n = 25) (B) to AZA. The heatmap of responding patients revealed several coordinated modules indicating a coordinated immune response, whereas no modules were formed in nonresponders when frequencies were clustered either spontaneously (i) or ordered from responders (ii). (C) No numerical differences in CD4+ T-cell subpopulations were observed between responders and nonresponders. (D) Adjacency matrix from panel A arranged by connectivity. Three CD4+ T-cell subpopulations emerged on the top 4 ranking positions. CD4+ T cells are in white; CD8+ T cells are in light blue.
CD4+T-cell subsets mediate coordination of the immune response in HR-MDS patients responding to AZA. (A-B) Pairwise correlations and hierarchical clustering of pretreatment peripheral blood (PB) and bone marrow (BM) blasts, absolute neutrophil count, and T-, and NK-cell frequencies in responders (n = 27) (A) and nonresponders (n = 25) (B) to AZA. The heatmap of responding patients revealed several coordinated modules indicating a coordinated immune response, whereas no modules were formed in nonresponders when frequencies were clustered either spontaneously (i) or ordered from responders (ii). (C) No numerical differences in CD4+ T-cell subpopulations were observed between responders and nonresponders. (D) Adjacency matrix from panel A arranged by connectivity. Three CD4+ T-cell subpopulations emerged on the top 4 ranking positions. CD4+ T cells are in white; CD8+ T cells are in light blue.
Transcriptomic analysis of CD4+ T cells reveals a differential regulation of gene expression depending on response to AZA
We further focused on CD4+ T cells and studied the molecular changes in bone marrow CD4+ T-cell populations during treatment with AZA. For this reason, we performed analysis of transcriptomic data from isolated cells from 4 responders and 4 nonresponders to AZA before the initiation and after 6 cycles (Cy6) of AZA administration (supplemental Table 3). Bioinformatic analysis identified 104 genes that were differentially expressed (false discovery rate [FDR] <0.2) in bone marrow CD4+ T cells from responders between Cy6 and pretreatment, whereas 165 genes were differentially expressed in nonresponders (Figure 2A-C). Ingenuity pathway analysis demonstrated that inflammatory and pathways involved in immune responses were overrepresented in the downregulated genes of CD4+ T cells from responders. In stark contrast, these pathways were overrepresented in the upregulated genes in CD4+ T cells from nonresponders (supplemental Figure 2). Genes involved in immune pathways were downregulated in responders after AZA administration, whereas IRF8, CD300E, CCL4, CCL3L3, CXCL8, and IL1B were upregulated in nonresponders. Gene set enrichment analysis (supplemental Figure 3) using the Molecular Signatures Database hallmark gene set collection showed a significant negative correlation of the transcriptomic signature of CD4+ T cells in responders with the inflammatory response and the IL6-JAK-STAT3 and the IL2-STAT5 signaling gene sets (Figure 2D). On the other hand, there was a significant positive correlation of the transcriptomic signature in nonresponders with the inflammatory response and the IL6-JAK-STAT3 signaling gene sets (Figure 2E). In addition, quantitative polymerase chain reaction analysis of STAT3 target genes in isolated CD4+ T cells from the peripheral blood of patients showed a trend toward downregulation of BCL3, BCL6, IL-21, KAT2B, and IRF8 in responders and upregulation of the same genes in nonresponders, although only alterations of BCL3 reached statistical significance (Figure 2F). Conversely, SOCS3, an inhibitor of STAT3 signaling and, in particular, of the IL-6/STAT3 pathway,25 exhibited a trend for upregulation in responders and downregulation in nonresponders, without, however, reaching statistical significance (supplemental Figure 4). Taken together, transcriptomic data in isolated CD4+ cells demonstrate an antithetic regulation of cytokine signaling by AZA between responders and nonresponders, in particular affecting modules of the JAK-STAT pathway.
![Effect of AZA treatment in the transcriptome of CD4+T cells from patients with HR-MDS. (A-B) Volcano plots depicting the differentially expressed genes in CD4+ T cells after 6 cycles of treatment compared with pretreatment in patients with complete response to treatment (CR, n = 4) (A) and in patients who failed AZA (n = 4) (B). The distribution of the FDR values (−log[FDR]) and the fold changes (log2 fold change) are shown. Significance level was adjusted to FDR <0.2. (C) Venn diagram depicting the number of differentially expressed genes after AZA therapy compared with pretreatment in patients with CR or failure to AZA. (D) Gene set enrichment analysis for genes associated with the inflammatory response, IL-6_JAK_STAT3 and IL-2_STAT5 signaling in patients with CR, and in patients that failed AZA (E). (F) Real-time quantitative polymerase chain reaction analysis of the STAT3 target gene BCL3 in isolated peripheral blood CD4+ T cells before and after 6 cycles of AZA (cy6). NES, normalized enrichment score.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/1/10.1182_bloodadvances.2020002351/3/m_advancesadv2020002351f2.png?Expires=1767698909&Signature=qapYTxHDJ6Rn63vB5yPnG9rmB3ny3XE7f4CN1DpWq~PlMIvA3iS9sJH-EKy4McIFDUYnh8v3McRN61e3p-6lXzRmvM1ki1HnrWrh4wQnfC~DbkXK8zTx7S3IfX9LxOsc90UO8vqQPV0~oL9il2B2QBd~KUXd8De2VwISd-erKoGSg195wtJe5BqGzOXFnbJ5dQI7Cw1JUs0BUr1G-DDseq20c9MfIdbE5YD1BN5c6pFyzFRPH57PbBDtn5sHTUVF0ZQqVk6Ndjqgg9rEfDsq7SoyFVmfGEgKJ54Ajvs102wnOLAtooivoQUfvDtbJ5oLb8CD1heX2sVveG1Ad0Seww__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of AZA treatment in the transcriptome of CD4+T cells from patients with HR-MDS. (A-B) Volcano plots depicting the differentially expressed genes in CD4+ T cells after 6 cycles of treatment compared with pretreatment in patients with complete response to treatment (CR, n = 4) (A) and in patients who failed AZA (n = 4) (B). The distribution of the FDR values (−log[FDR]) and the fold changes (log2 fold change) are shown. Significance level was adjusted to FDR <0.2. (C) Venn diagram depicting the number of differentially expressed genes after AZA therapy compared with pretreatment in patients with CR or failure to AZA. (D) Gene set enrichment analysis for genes associated with the inflammatory response, IL-6_JAK_STAT3 and IL-2_STAT5 signaling in patients with CR, and in patients that failed AZA (E). (F) Real-time quantitative polymerase chain reaction analysis of the STAT3 target gene BCL3 in isolated peripheral blood CD4+ T cells before and after 6 cycles of AZA (cy6). NES, normalized enrichment score.
Effect of AZA treatment in the transcriptome of CD4+T cells from patients with HR-MDS. (A-B) Volcano plots depicting the differentially expressed genes in CD4+ T cells after 6 cycles of treatment compared with pretreatment in patients with complete response to treatment (CR, n = 4) (A) and in patients who failed AZA (n = 4) (B). The distribution of the FDR values (−log[FDR]) and the fold changes (log2 fold change) are shown. Significance level was adjusted to FDR <0.2. (C) Venn diagram depicting the number of differentially expressed genes after AZA therapy compared with pretreatment in patients with CR or failure to AZA. (D) Gene set enrichment analysis for genes associated with the inflammatory response, IL-6_JAK_STAT3 and IL-2_STAT5 signaling in patients with CR, and in patients that failed AZA (E). (F) Real-time quantitative polymerase chain reaction analysis of the STAT3 target gene BCL3 in isolated peripheral blood CD4+ T cells before and after 6 cycles of AZA (cy6). NES, normalized enrichment score.
Downregulation of IL-6–induced STAT3 phosphorylation in Tcons after AZA administration is associated with better disease outcome
Prompted by the downregulation of the IL6-JAK-STAT3 and IL2-STAT5 signaling pathways in BM CD4+ T cells on treatment with AZA in responders, we further studied the STAT signaling profile of the peripheral blood CD4+FOXP3− Tcons and CD4+FOXP3+ T cells (Tregs) by multiparametric phospho-flow cytometry. We observed that patients who decreased the IL-6–induced STAT3 phosphorylation in Tcons (n = 28, IL-6/STAT3_down) after 6 cycles of treatment with AZA displayed significant longer OS (P = .006) and EFS (P = .015) compared with patients whose Tcons upregulated the IL-6/STAT3 node after treatment with AZA (n = 26, IL-6/STAT3_up group; Figure 3A-B). Consistent with these results, patients in the IL-6/STAT3_down group also showed a markedly better response to AZA compared with the IL-6/STAT3_up group (P = .001; Table 1; Figure 3C). Multivariate analysis further confirmed the independent predictive value of IL-6/STAT3 pathway modifications on patient outcome (supplemental Table 4; Figure 3D-E). In contrast, no correlations of AZA-induced IL-2/STAT5 axis alterations in Tcons were observed with OS, EFS, or response to therapy (supplemental Figure 5). Likewise, AZA-induced modifications of either IL-6/STAT3 or IL-2/STAT5 pathways in Tregs were not associated with response and survival (supplemental Figure 6Α-Β).
![Downregulation of IL-6–induced STAT3 phosphorylation in CD4+FOXP3−Tcons after AZA correlates significantly with better response and survival of HR-MDS patients. Kaplan-Meier curves for OS (A) and EFS (B) in HR-MDS patients who down- or upregulated the IL-6/STAT3 node in Tcons. Median OS of downregulators of IL-6/STAT3 (n = 28) was 27.2 months (95% confidence interval [CI]: 15-39.4) compared with 11.2 months (95% CI: 9.1-13) for upregulators (n = 26) (hazard ratio [HR]: 0.41, 95% CI: 0.22-0.78, P = .006). Median EFS of downregulators of IL-6/STAT3 was 16.6 months (95% CI: 8.1-25) compared with 9.45 months (95% CI: 7.55-11.05)] for upregulators (HR: 0.47, 95% CI: 0.25-0.86, P = .015). (C-D) Likewise, IL-6/STAT3 downregulators displayed significantly higher response rates (C), whereas no differences were noted between the 2 groups regarding World Health Organization subtype, cytogenetics according to IPSS-R and IPSS-R category, and transfusion intensity (D). (E) Forest plot of univariate analysis of predictive factors for OS. Upregulation of the IL-6/STAT3 node in Tcons was the only significant parameter in multivariate analysis. IPSS-R, Revised International Prognostic Scoring System.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/1/10.1182_bloodadvances.2020002351/3/m_advancesadv2020002351f3.png?Expires=1767698909&Signature=WzyWUibBIrZkrvMsDsLaaxdyiFdlsQjeUUk9vp0LDBRQrSwXmveE2oB8g-Y2kAyxWsGFIX-gW-sBPMYJSjHkE6Q2sTF2G4ygUYvYXVmqelTUyE-k-PJiCtmbaLQieY-woiySmUhyucDhnwFhujqE35g6tg0OnQs1Z~rz1Pp-eyW1qiPp~qcHNK77cJsnIk1d0aYk9vioA3CB~PVr50Rsoyo1WBBfB5F0zfuKKBKySbXZ~BX9wd9Yvere4GRM9CG87e6IvQXr59jTqhpVc6H6L4fNwQRietW28JNpTY91pIK8e2IWQceTCITcTa0z3likLwIdaBTmJ~cw1VZK0~YKmg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Downregulation of IL-6–induced STAT3 phosphorylation in CD4+FOXP3−Tcons after AZA correlates significantly with better response and survival of HR-MDS patients. Kaplan-Meier curves for OS (A) and EFS (B) in HR-MDS patients who down- or upregulated the IL-6/STAT3 node in Tcons. Median OS of downregulators of IL-6/STAT3 (n = 28) was 27.2 months (95% confidence interval [CI]: 15-39.4) compared with 11.2 months (95% CI: 9.1-13) for upregulators (n = 26) (hazard ratio [HR]: 0.41, 95% CI: 0.22-0.78, P = .006). Median EFS of downregulators of IL-6/STAT3 was 16.6 months (95% CI: 8.1-25) compared with 9.45 months (95% CI: 7.55-11.05)] for upregulators (HR: 0.47, 95% CI: 0.25-0.86, P = .015). (C-D) Likewise, IL-6/STAT3 downregulators displayed significantly higher response rates (C), whereas no differences were noted between the 2 groups regarding World Health Organization subtype, cytogenetics according to IPSS-R and IPSS-R category, and transfusion intensity (D). (E) Forest plot of univariate analysis of predictive factors for OS. Upregulation of the IL-6/STAT3 node in Tcons was the only significant parameter in multivariate analysis. IPSS-R, Revised International Prognostic Scoring System.
Downregulation of IL-6–induced STAT3 phosphorylation in CD4+FOXP3−Tcons after AZA correlates significantly with better response and survival of HR-MDS patients. Kaplan-Meier curves for OS (A) and EFS (B) in HR-MDS patients who down- or upregulated the IL-6/STAT3 node in Tcons. Median OS of downregulators of IL-6/STAT3 (n = 28) was 27.2 months (95% confidence interval [CI]: 15-39.4) compared with 11.2 months (95% CI: 9.1-13) for upregulators (n = 26) (hazard ratio [HR]: 0.41, 95% CI: 0.22-0.78, P = .006). Median EFS of downregulators of IL-6/STAT3 was 16.6 months (95% CI: 8.1-25) compared with 9.45 months (95% CI: 7.55-11.05)] for upregulators (HR: 0.47, 95% CI: 0.25-0.86, P = .015). (C-D) Likewise, IL-6/STAT3 downregulators displayed significantly higher response rates (C), whereas no differences were noted between the 2 groups regarding World Health Organization subtype, cytogenetics according to IPSS-R and IPSS-R category, and transfusion intensity (D). (E) Forest plot of univariate analysis of predictive factors for OS. Upregulation of the IL-6/STAT3 node in Tcons was the only significant parameter in multivariate analysis. IPSS-R, Revised International Prognostic Scoring System.
To address whether the modulation of IL-6/STAT3 axis in Tcons is induced by AZA itself or it is a mere corollary of immune system reshaping because of the clearance of the leukemic clone, we measured IL-6–dependent STAT3 phosphorylation in Tcons at day 15 after AZA initiation when leukemic burden is still high. Patients who achieved CR significantly downregulated the IL-6/STAT3 node in Tcons (n = 6) at day 15 of the first AZA cycle (P = .02), whereas there was no difference in STAT3 phosphorylation in patients who failed AZA (n = 9, P = .85), supporting a direct effect of AZA on the IL-6/STAT3 signaling pathway in Tcons of responding patients (Figure 4A-B).

Modulation of IL-6/STAT3 axis in Tcons is mediated by AZA and is independent of IL-6 receptor levels. (A) Representative flow cytometry histograms of a patient with CR and a patient who failed AZA gated on Tcons. (B) A significant downregulation of the IL-6/STAT3 node was already evident at day 15 of the first AZA cycle in patients who achieved CR (n = 6), whereas there was no modification in STAT3 phosphorylation in patients who failed treatment (n = 9). Data shown as mean ± standard error of the mean. (C) No differences in the expression of IL-6Rα in Tcons were observed between downregulators (n = 5) or upregulators (n = 5) either prior (P = .54) or after AZA (P = .94). Likewise, AZA therapy did not modify the expression of IL-6Rα in Tcons in both downregulators (P = .1) and upregulators (P = .18; i). Similar to IL-6Rα, gp130 expression was comparable between downregulators and upregulators both before (P = .22) and after AZA (P = .66), whereas AZA therapy did not alter significantly gp130 levels in Tcons in both downregulators (P = .31) and upregulators (P = .31; ii). *P < .05. CR, complete remission; UT, untreated/unmodulated node; NMFI, normalized median fluorescence intensity.
Modulation of IL-6/STAT3 axis in Tcons is mediated by AZA and is independent of IL-6 receptor levels. (A) Representative flow cytometry histograms of a patient with CR and a patient who failed AZA gated on Tcons. (B) A significant downregulation of the IL-6/STAT3 node was already evident at day 15 of the first AZA cycle in patients who achieved CR (n = 6), whereas there was no modification in STAT3 phosphorylation in patients who failed treatment (n = 9). Data shown as mean ± standard error of the mean. (C) No differences in the expression of IL-6Rα in Tcons were observed between downregulators (n = 5) or upregulators (n = 5) either prior (P = .54) or after AZA (P = .94). Likewise, AZA therapy did not modify the expression of IL-6Rα in Tcons in both downregulators (P = .1) and upregulators (P = .18; i). Similar to IL-6Rα, gp130 expression was comparable between downregulators and upregulators both before (P = .22) and after AZA (P = .66), whereas AZA therapy did not alter significantly gp130 levels in Tcons in both downregulators (P = .31) and upregulators (P = .31; ii). *P < .05. CR, complete remission; UT, untreated/unmodulated node; NMFI, normalized median fluorescence intensity.
The AZA-mediated IL-6–induced STAT3 phosphorylation in Tcons is not associated with IL-6 cytokine and receptor levels
We then addressed whether the alterations of IL-6–induced STAT3 phosphorylation in Tcons after AZA were caused by changes in either IL-6 levels or the expression of IL-6 receptors. No differences in the surface expression of IL-6Rα and gp130 in Tcons were noted between IL-6/STAT3_down (n = 5) and __up (n = 5) patients either before (P = .54 and P = .22, respectively, for IL-6Rα and gp130) or after (P = .94 and P = .66) AZA. Also, expression of IL-6Rα and gp130 in Tcons remained unchanged after 6 cycles of AZA in both IL-6/STAT3_down (P = .1 and P = .31) and IL-6/STAT3_up patients (P = .18 and P = .31) (Figure 4C), further indicating that the AZA-mediated differential regulation of IL-6/pSTAT3 axis in Tcons is not caused by alterations in IL-6 receptor levels. As IL-6 can use the alternative pathway of trans-signaling by forming a complex with the soluble IL-6 receptor (sIL-6R),26 we also assessed the bone marrow plasma levels of sIL-6R. Both sIL-6R and IL-6 levels followed the same trajectory, irrespective of the AZA-mediated IL-6/STAT3 modifications in Tcons (supplemental Figure 7). However, as only 2 upregulators had available measurements, no formal statistical analysis could be done, and a definitive conclusion cannot be drawn.
Alterations of IL-6–induced STAT3 phosphorylation in Tcons by AZA are not associated with the mutation number and profile of HR-MDS patients
Despite the reported connection between the tumor mutational and immune profiles in solid tumors,27 data on myeloid malignancies are limited, and the interrelation of cellular immunity with somatic mutations is largely unclear.28 We sought to investigate whether the modifications of IL-6–induced pSTAT3 phosphorylation in Tcons by AZA correlate with the mutational and cytogenetic alterations of the patients. The distribution of mutations in down- and upregulators is depicted in Figure 5A. The number of oncogenic mutations before treatment was comparable among IL-6/STAT3_down and IL-6/STAT3_up patients (P = .73; Figure 5B), whereas categorization of oncogenic mutations as previously described29 revealed no associations with the AZA-induced IL6/STAT3 signaling alterations in Tcons (Figure 5C). Likewise, no other single mutation and/or karyotypic abnormality correlated with IL6/STAT3 changes in Tcons after AZA (Figure 5D). Comutation analysis in IL-6/STAT3_down or _up groups also found no significant associations, but the comutation patterns within groups were heavily underpowered because of the limited size of our cohort, thus precluding any definite conclusions (data not shown).

AZA-mediated modulations of IL-6–induced STAT3 phosphorylation in Tcons are not linked to the mutation number and profile of HR-MDS patients. (A) Spectrum of recurrent mutations and cytogenetic abnormalities in patients who downregulated (green) or upregulated (purple) the IL-6/STAT3 signaling axis in Tcons. Each column represents an individual patient sample. (Β) Comparable number of oncogenic mutations before AZA initiation between IL-6/STAT3 downregulators and upregulators. (C) No associations of any functional pathway of oncogenic mutations with the AZA-induced IL6/STAT3 signaling alterations in Tcons. (D) Frequency of driver mutations or karyotypic abnormalities in the IL-6/pSTAT3_down (green) and IL-6/pSTA3_up (blue) groups. No statistically significant associations of single mutations with IL-6/STAT3 modifications were found.
AZA-mediated modulations of IL-6–induced STAT3 phosphorylation in Tcons are not linked to the mutation number and profile of HR-MDS patients. (A) Spectrum of recurrent mutations and cytogenetic abnormalities in patients who downregulated (green) or upregulated (purple) the IL-6/STAT3 signaling axis in Tcons. Each column represents an individual patient sample. (Β) Comparable number of oncogenic mutations before AZA initiation between IL-6/STAT3 downregulators and upregulators. (C) No associations of any functional pathway of oncogenic mutations with the AZA-induced IL6/STAT3 signaling alterations in Tcons. (D) Frequency of driver mutations or karyotypic abnormalities in the IL-6/pSTAT3_down (green) and IL-6/pSTA3_up (blue) groups. No statistically significant associations of single mutations with IL-6/STAT3 modifications were found.
Downregulation of IL-6/STAT3 activity in Tcons restores the STAT signaling architecture in Tcons and Tregs
To address the effect of AZA-mediated modifications of IL-6/STAT3 axis in Tcons on the T cell–mediated immune response, we first assessed the peripheral blood levels of various T-cell subsets before treatment and after 6 cycles of AZA administration. We only observed a significant increase in the frequency of both PD1+CD4+ (P = .02) and PD1+CD8+ (P = .03) T cells at cycle 6 in the IL-6/STAT3_up group. No changes were noticed in the rest of the CD4+ and CD8+ T- and NK-cell subsets in both patient groups (supplemental Figures 8 and 9). We further performed pairwise correlations of immune subsets and STAT signaling nodes within and among each of Tcons and Tregs before treatment initiation at day 15 and at Cy6. Hierarchical clustering of correlations of immune cell frequencies revealed several coordinated modules both before and after AZA therapy in the IL-6/STAT3_down group, whereas no modules were formed in nonresponders (supplemental Figure 10). Importantly, the STAT signaling network map uncovered a completely disorganized STAT signaling architecture of Tcons and Tregs in both the IL-6/STAT3_down and IL-6/STAT3_up groups before AZA administration and also at day 15 after the first cycle. Downregulation of IL-6–dependent STAT3 phosphorylation in Tcons after 6 cycles of AZA was accompanied by the restoration of a high degree of connectivity between cytokinic responses in Tcons and Tregs (Figure 6A). By contrast, in IL-6/STAT3_up patients, the STAT signaling network remained deranged in Tcons and Tregs (Figure 6B). Thus, it appears that the AZA-induced downregulation of IL-6/STAT3 activity in Tcons gradually reorganizes the STAT signaling network in Tcons and Tregs.
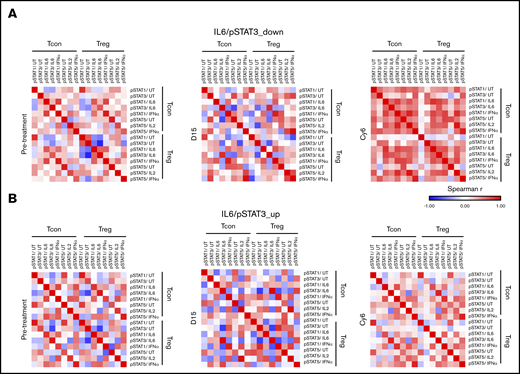
Downregulation of IL-6/STAT3 activity in Tcons reestablishes the connectivity of STAT signaling network in Tcons and Tregs. Functional maps of STAT signaling network generated by calculating Spearman’s rank correlation coefficients between pairs of nodes within and among each of Tregs and Tcons before initiation and after 6 cycles of AZA. (A) Although the STAT architecture was completely disorganized in all patients both before and 15 days (D15) after AZA initiation, the AZA-mediated downregulation of IL-6/STAT3 signaling in Tcons was accompanied by restructuring of STAT networks and the formation of numerous positively correlated nodes across Tcons and Tregs on cycle 6 (Cy6). (B) By contrast, the pattern of signaling responses remain disorganized in patients who upregulated IL-6/STAT3 signaling in Tcons after 6 cycles of AZA.
Downregulation of IL-6/STAT3 activity in Tcons reestablishes the connectivity of STAT signaling network in Tcons and Tregs. Functional maps of STAT signaling network generated by calculating Spearman’s rank correlation coefficients between pairs of nodes within and among each of Tregs and Tcons before initiation and after 6 cycles of AZA. (A) Although the STAT architecture was completely disorganized in all patients both before and 15 days (D15) after AZA initiation, the AZA-mediated downregulation of IL-6/STAT3 signaling in Tcons was accompanied by restructuring of STAT networks and the formation of numerous positively correlated nodes across Tcons and Tregs on cycle 6 (Cy6). (B) By contrast, the pattern of signaling responses remain disorganized in patients who upregulated IL-6/STAT3 signaling in Tcons after 6 cycles of AZA.
Discussion
Despite its extensive use in MDS and acute myeloid leukemia (AML), the exact mechanism of action and the genetic and cellular levels where AZA acts are yet unidentified,30 whereas resistance to AZA appears to be multifactorial, involving pharmacologic31,32 and molecular33 processes. AZA may promote the antitumor immune response by several mechanisms encompassing increased tumor immunogenicity and enhanced cellular and cytokine-mediated effector T-cell tumor lysis.34-36 However, there are currently limited data on the role of CD4+ T cells, an essential component of tumor immunity, in the immune landscape of HR-MDS and the antitumor immune response elicited by AZA. In the present work, we show that CD4+ T cells hold a central role in the coordination of the immune response in HR-MDS and are differentially affected by AZA depending on treatment response. Importantly, we identified an AZA-mediated, prognostically favorable, downregulation of the IL-6/STAT3 signaling axis in CD4+FOXP3− T cells, thus revealing a novel beneficial immune-mediated effect of AZA.
Pairwise correlations of leukemic and immune cells revealed the existence of a coordinated immune response only in responders to AZA and pinpointed CD4+ T cells as a key regulator of cellular immunity in HR-MDS. These findings bear a remarkable resemblance to murine tumor models in which CD4+ T cell–mediated coordination of the immune response was crucial for tumor remission after immunotherapy,37 further arguing for the critical role of CD4+ cells in the antineoplastic effect mediated by AZA.
We previously showed that the transcriptional profile of CD4+ T cells in MDS and chronic myelomonocytic leukemia is indistinguishable but differs markedly with the one of normal individuals and patients with de novo AML.38 We now report a diverse regulation of CD4+ T-cell transcriptome depending on the response to AZA. Inflammatory and cytokine signaling pathways were downregulated in responders, whereas AZA failure was accompanied by the emergence of a robust inflammatory transcriptomic signature. In particular, the IL6/STAT3 and the IL2/STAT5 signaling axes were mostly affected by AZA, and using functional phenotyping, we found that AZA-mediated downregulation of the IL-6/STAT3 node in CD4+FOXP3− Tcons was a powerful predictor of better treatment response and outcome. Of note, remission duration was also significantly longer in IL-6/STAT3 downregulators, potentially reflecting a more effective immune-mediated control of the leukemic clone in these patients (supplemental Figure 11). Moreover, clinical and biological characteristics were similar between patients who down- or upregulated IL-6/STAT3 signaling, underscoring the independent prognostic information that can be obtained by single-cell STAT network profiling in both leukemic21,39,40 and immune cells.
In patients with breast cancer, defective pretreatment IL-6–induced STAT3 phosphorylation in peripheral blood–naïve CD4+ T cells correlated with higher risk of relapse, whereas patients who achieved remission normalized the levels of the IL-6/STAT3 node.41 In contrast, we did not find any associations of pretreatment IL-6/STAT3 levels in any T-cell subset with treatment response (data not shown). We speculate that the diverse underlying pathobiology between solid tumors and myeloid clonal neoplasms might account for this discrepancy. In this respect, although principally protumorigenic, IL-6/STAT3 signaling in certain circumstances may operate to boost antitumor immunity.42 In the same study, the blunted stimulation of STAT3 by IL-6 was attributed to decreased expression of the IL-6 coreceptors, gp130 and IL-6Rα, in naïve CD4+ T cells. In contrast, we were not able to identify any significant associations of alterations in IL-6/STAT3 signaling with bone marrow plasma levels of IL-6 and the expression of both membrane and soluble IL-6 coreceptors. The dissociation between receptor levels and STAT3 phosphorylation was also reported in normal and AML CD34+ cells,21,39,43 indicating that qualitative abnormalities of IL-6 coreceptors or receptor-associated proteins, dysfunctional downstream signaling elements, or alterations of positive or negative regulators of IL-6/STAT3 signaling axis are potentially responsible for the disparity in IL-6 sensitivity of Tcons in our patients.
The mutational profile of neoplastic cells may sculpt specific immune response patterns, common across heterogeneous tumors.27 In AML, various mutations affect extrinsically T cell–mediated antitumor immunity,44 but it is still unknown if driver mutations in T cells beget an intrinsic defect in T-cell immunity.28 The AZA-induced IL6/STAT3 alterations in Tcons were not associated with either cytogenetics or the number and profile of somatic mutations in HR-MDS patients. However, given the vast genetic variability of MDS and the still unknown implications of comutation patterns in antitumor immunity, considerably larger patient cohorts are needed to reliably assess for a causal link between genetic alterations and the AZA-mediated modulation of IL-6/STAT3 axis in Tcons.
Aberrant IL-6 signaling via STAT3 in CD4+ T cells has profound effects on antitumor immunity by encompassing a variety of mechanisms, such as intratumoral expansion of immunosuppressive Tregs and suppression of antitumor Th1 responses.45,46 In our cohort, we only observed a significant increase in both PD1+CD4+ and PD1+CD8+ T-cell levels after 6 cycles of AZA in IL-6/STAT3 upregulators, consistent with a report showing that the AZA-mediated induction of PD-1 in T cells correlates with refractory disease.47 Nevertheless, as mere arithmetical changes of T-cell subpopulations cannot uncover the complex interplay between interdependent immune cell subsets and signaling pathways, we assessed at the single-cell level, the functional capacity of the STAT signaling network by multiparametric phosphor-specific cytometry. The downregulation of IL-6/STAT3 axis in Tcons led to a high degree of connectivity of intra- and intersubset cytokine signaling in Tcons and Tregs after AZA, whereas the STAT cell signaling network remained disorganized in patients that upregulated the IL-6/STAT3 node. Because coordinated responses to cytokines across immune cells is a characteristic of normal immunity,48 our findings indicate that AZA reinstates the STAT signaling architecture of CD4+ cells in downregulators of the IL-6/STAT3 axis in Tcons and, along with the AZA-mediated transcriptomic changes, point to a direct cell-specific effect on CD4+ T cells. The striking similarity with our previous work showing an advantageous AZA-mediated downregulation of STAT3/5 signaling in CD34+ progenitors21 further argues that AZA directly affects signaling networks on specific cell types. Although no clinically relevant alterations of IL-6/STAT3 pathway were found in other CD4+ T-cell subsets, the fact that diverse cytokine stimuli in diverse cell types converge to and alter STAT3 signaling suggests that this pathway is preferably targeted by AZA in pivotal subsets of both immune and myeloid cells. Consistent with this, in 18 patients with available data on both CD34+ and CD4+ cells, AZA-mediated STAT3 signaling alterations in Tcons and CD34+ cells were almost always at the same direction in each individual patient (supplemental Figure 12). In summary, our study puts forward a previously unrecognized role of CD4+ T cells in the immune mechanisms used by AZA. The prognostically relevant AZA-mediated downregulation of IL-6/STAT3 signaling in Tcons reestablishes a close to normal STAT signaling network in CD4+ T cells and potentially subserves the antileukemic immune response. The IL-6/STAT3 signaling axis is notoriously protumorigenic, and pharmacologic inhibition of various individual modules of this pathway in cancer is under development.45 Along with the effects of AZA on the STAT biosignature of clonal CD34+ cells,21 our findings help pave the way for the investigation of IL-6/STAT3 axis inhibition as novel approach to overcome AZA resistance in HR-MDS. In this sense, successful cotargeting of IL-6/STAT3 signaling and immune checkpoint molecules in murine tumor models49,50 argues further for the development of rational AZA combinations based on patient-specific STAT biosignatures.
RNA-seq data were previously published and deposited in the EMBL-EBI repository (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-8208.
Acknowledgments
The authors thank Haralampos Hatzikirou for input in the formation of adjacency matrix, Vasileios Papadopoulos for input in the statistical analysis, and Emmanuil Spanoudakis and Katerina Chlichlia for providing important intellectual assistance.
This study was supported in part by a research grant by Celgene (NCRG-GRC-056). I.M. was supported by the Hellenic Foundation for Research and Innovation (grant 452) and General Secretariat for Research and Technology Management and Implementation Authority for Research, Technological Development and Innovation Actions (MIA-RTDI) (grant Τ2EDK-02288, MDS-TARGET). C.K. is supported by Greece and the European Union European Social Fund (ESF) through the operational programme “Human Resources Development, Education and Lifelong Learning,” in the context of the project “Strengthening Human Resources Research Potential via Doctorate Research” (MIS-5000432), implemented by the State Scholarships Foundation.
Authorship
Contribution: I.K., I.M., and E.L. designed, performed, and analyzed the experiments, performed the statistical analysis, and wrote the article; C.K. and E.Z. designed and analyzed the experiments; G.K., A.K., A.F., and P.G. performed and analyzed the transcriptional profiling; E.P., E.B., S.L., and A.C. performed and analyzed next-generation sequencing; I.K., S.G.P., T.V.P., A.G.G., N.A.V., and V.P. enrolled patients and collected and annotated patient data; E.N. and L.K. performed flow cytometry experiments and immunoassays; and all authors discussed the results and commented on the manuscript.
Conflict-of-interest disclosure: I.K. and V.P. received research funding from Celgene Corporation and honoraria from Genesis Pharma Hellas. S.G.P., T.V.P., A.G.G., and N.A.V. received honoraria from Genesis Pharma Hellas. The remaining authors declare no competing financial interests.
Correspondence: Ioannis Kotsianidis, Department of Hematology, Democritus University of Thrace, Medical School, Dragana, Alexandroupolis 68100, Greece; e-mail: ikotsian@med.duth.gr.

